A Complete Guide to Subculturing Adherent Insect Cells: Protocols, Troubleshooting, and Best Practices
This article provides a comprehensive guide for researchers and drug development professionals on the subculture of adherent insect cells, a critical technique for recombinant protein production and biotherapeutics.
A Complete Guide to Subculturing Adherent Insect Cells: Protocols, Troubleshooting, and Best Practices
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on the subculture of adherent insect cells, a critical technique for recombinant protein production and biotherapeutics. It covers the foundational principles of insect cell biology, offers step-by-step methodological protocols for Sf9 and Sf21 cell lines, details advanced troubleshooting and optimization strategies to maintain high viability, and includes a comparative analysis with mammalian and suspension systems. The content synthesizes current best practices to ensure robust, reproducible, and scalable cell culture processes for biomedical applications.
Understanding Adherent Insect Cells: Biology and Significance in Bioproduction
Defining Adherent Insect Cell Culture and Anchorage Dependence
Within the broader scope of research on subculturing adherent insect cells, understanding the defining characteristic of anchorage dependence is fundamental. Adherent insect cell culture describes the in vitro growth of insect cells that require attachment to a solid, growth-promoting substrate for proliferation and survival [1] [2]. This requirement for attachment is termed anchorage dependence, a trait common to all normal, non-transformed cells derived from tissues other than the hematopoietic system [2]. In contrast, transformed or tumor-derived cells can often proliferate in suspension without a surface [2]. For insect cells, this dependence dictates specific culture conditions, subculture protocols, and scalable production strategies, making it a critical concept in applications ranging from viral pesticide production and baculovirus expression vector systems to the development of novel vaccines [2] [3]. This document outlines the core biological principles and provides detailed, actionable protocols for the effective culture and subculturing of adherent insect cells.
Biological Principles of Anchorage Dependence
Core Concept and Cellular Mechanisms
Anchorage dependence is a biological imperative for many cell types. Normal cells, including most primary insect cells, integrate signals from the extracellular matrix (ECM) through integrins to regulate their cell cycle [2]. The absence of this attachment sends a signal for growth arrest and can induce a specific form of programmed cell death known as anoikis [2]. Essentially, without a surface to adhere to, anchorage-dependent cells cannot sense a proper environment for division and will initiate self-destruction pathways. For insect cells, the interaction with the substrate is mediated by cellular integrins and is crucial for self-renewal, expansion, and the maintenance of a particular phenotype [2].
Distinction from Other Cell Types
The property of anchorage dependence allows for a clear classification of cell cultures, which is essential for selecting the appropriate cultivation platform. The table below summarizes the key differences.
Table 1: Classification of Cell Culture Types Based on Anchorage Dependence
| Cell Type | Anchorage Requirement | Typical Morphology in Culture | Examples | Key Growth Characteristics |
|---|---|---|---|---|
| Adherent | Requires attachment to a substrate | Forms a monolayer on the surface | Vero, MRC-5, HEK293, many primary insect cells [1] [2] [4] | Anchorage-dependent; proliferation ceases if detached [2] |
| Suspension | No attachment required | Grows freely floating in the medium | BHK21, EB66, adapted CHO cells [2] | Anchorage-independent; can be cultured in stirred-tank reactors [4] |
| Semi-adherent | Loosely attached | Mixed population: some adherent, some in suspension | Some insect cell lines (e.g., Trichoplusia ni TN-368) [5] | May require gentle methods to dislodge adherent fraction [5] |
Materials and Reagents for Adherent Insect Cell Culture
Successful culture relies on a defined set of reagents and equipment tailored to the unique needs of insect cells.
Table 2: Essential Research Reagent Solutions for Adherent Insect Cell Culture
| Reagent/Equipment | Function/Application | Specific Examples & Notes |
|---|---|---|
| Culture Media | Provides nutrients, pH buffering, and osmotic balance. | Grace's Insect Medium, Schneider's Drosophila Medium [3]. Formulations are order-specific (Lepidoptera, Diptera) [3]. |
| Dissociation Reagent | Detaches adherent cells from the culture surface for subculturing. | Trypsin (e.g., VMF trypsin) [5] [3]. For strongly adherent cells, physical methods may be preferred to avoid enzymatic damage [5]. |
| Cryoprotectant | Protects cells from ice crystal formation during freezing. | DMSO (5-10%) or glycerol (2-20%) in serum-containing media [4]. |
| Serum/Supplements | Provides growth factors and protects from shear stress. | Fetal Bovine Serum (FBS) or serum-free formulations containing surfactants like Pluronic F-68 [3]. |
| Culture Vessels | Provides a sterile, treated surface for cell attachment and growth. | 25-cm² or 75-cm² tissue culture flasks [5] [3]. |
| Incubator | Maintains optimal temperature and, if needed, humidity. | Set to 27-28°C; non-humidified; CO₂ is not required [1] [3]. |
Protocols for Subculturing Adherent Insect Cells
The following protocols are critical for maintaining healthy, expanding cell populations. Adherence to aseptic technique is assumed throughout all procedures.
Protocol 1: Subculturing Loosely Adherent and Non-Attached Insect Cells
This method is suitable for cell lines that grow in suspension or attach weakly, such as Trichoplusia ni TN-368 and Lymantria dispar IPLB-LdFB [5].
Detailed Methodology:
- Preparation: Examine the mature culture under an inverted phase-contrast microscope. The medium should be clear, and cells should appear refractive, indicating health. Cloudiness suggests contamination [5].
- Labeling: Label new culture flasks with the cell line designation, passage number, and date [5].
- Medium Addition: Aseptically add a predetermined volume of fresh, pre-warmed culture medium to the new flask(s). For a 1:10 split ratio in a 25 cm² flask, add 3.6 mL of fresh medium [5].
- Cell Harvesting: Gently swirl the mature culture to evenly disperse the cells. Using a sterile pipette, draw up the appropriate volume of cell suspension (0.4 mL for a 1:10 split) [5].
- Inoculation: Dispense the cell suspension into the new flask containing fresh medium [5].
- Incubation: Tighten the cap and place the new culture in a 27°C incubator. CO₂ exchange and humidification are not required [1] [3].
The workflow for this protocol is outlined below.
Protocol 2: Subculturing Strongly Adherent Insect Cells
This protocol is designed for firmly attached cell lines like Spodoptera frugiperda Sf-9, Sf-21, and Manduca sexta MRRL-CH1 [5].
Detailed Methodology:
- Cooling: Place the mature culture in a 4°C refrigerator for 15-20 minutes. This step depolymerizes cell microtubules, weakening adhesion [5] [3].
- Cell Detachment: Remove the culture from refrigeration. Firmly tap the side of the flask with the palm of your hand 2-3 times to dislodge the cells. For some lines, vigorous flushing with culture medium from a pipette may be necessary [5].
- Completion: Continue from Step 3 of Protocol 1, adding fresh medium to new flasks and transferring the detached cell suspension.
Table 3: Troubleshooting Common Issues in Subculturing
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| Poor Detachment | Incorrect confluency at passaging; insufficient force. | For strongly adherent cells, passage at confluency or slightly after, when cells may start to pull away [1]. Ensure firm, sharp tapping. |
| Decreased Doubling Time / Viability | Repeatedly passaging at incorrect density. | Avoid passaging at densities past confluency or too low (<20% confluency). Find the optimal passaging density for the specific cell line [1]. |
| Cell Clumping After Subculture | Overly aggressive dissociation; cell damage. | Avoid vigorous shaking. Use controlled, sharp taps. If using trypsin, neutralize promptly with serum-containing medium [5] [3]. |
Scaling Up Adherent Insect Cell Culture
For industrial applications in vaccine production or biotherapeutics, moving beyond flask-based systems is necessary. Scaling up adherent cultures requires increasing the available surface area for cell attachment.
Table 4: Scalable Culture Systems for Anchorage-Dependent Cells
| Production Technology | Principle | Largest Scale Available | Applications |
|---|---|---|---|
| Roller Bottles | Cylindical bottles rotated to alternately cover cells with medium and expose to air. | Up to 734 m² per system [2] | Veterinary vaccines, EPO production [2] |
| Cell Factories / Stacks | Multilayered polystyrene stacks providing a large surface area in a compact footprint. | Up to 25,280 cm² per 40-stack unit [2] | Beta-interferon production, vaccine R&D [2] |
| Microcarriers in Stirred-Tank Reactors | Cells grow on small beads suspended in a bioreactor via agitation. | 6000 L (est. 2430 m² surface) [2] | Large-scale human vaccine production (e.g., influenza, polio) [2] |
| Packed-Bed Bioreactors (e.g., Tide Motion) | Cells attach to stationary macroporous carriers; media is perfused in a "tide motion". | Up to 100 L packed-bed (equiv. to >100,000 flasks) [6] | Vaccine and viral production, stem cell therapy [6] |
The Tide Motion technology, a modern packed-bed approach, creates a low-shear environment ideal for sensitive insect cells. The following diagram illustrates its core principle.
Tide Motion Bioreactor Cycle
The defining feature of adherent insect cell culture is anchorage dependence, a biological imperative that directly shapes all subsequent protocols and scale-up strategies. Mastery of the specific subculturing techniques—whether for loosely or strongly adherent cell lines—is fundamental to successful research and development. As the field advances towards industrial-scale production of viruses, recombinant proteins, and novel biologics, leveraging scalable technologies like microcarriers and packed-bed bioreactors becomes essential. Understanding these principles and protocols ensures that researchers can effectively maintain the health and functionality of adherent insect cells, thereby underpinning robust and reproducible scientific and commercial outcomes.
Insect cell lines are a cornerstone of modern bioprocessing, providing a versatile platform for recombinant protein production, vaccine development, and biomedical research. Within this field, the Sf9, Sf21, and High-Five (BTI-Tn-5B1-4) cell lines have emerged as the most widely utilized systems, particularly when paired with the baculovirus expression vector system (BEVS). Their ability to perform complex post-translational modifications, coupled with high protein yields and scalability, makes them indispensable in both academic and industrial settings. This application note details the origins, key characteristics, and practical protocols for maintaining these cell lines, providing a essential guide for researchers within the broader context of subculturing adherent insect cells.
Origins and Lineage
The development of these cell lines is rooted in decades of entomological and biotechnological research.
- Sf21: The IPLB-Sf-21 cell line was established in 1977 from the pupal ovarian tissue of the fall armyworm, Spodoptera frugiperda [7] [8]. It was noted for its high susceptibility to baculovirus infection, making it an immediate candidate for recombinant protein production.
- Sf9: The Sf9 cell line is a clonal isolate derived from the parental Sf21 cell line [9] [10] [11]. This clonal selection resulted in a population with a more uniform, regular size and morphology, which has made it the cell line of choice in many laboratories for working with recombinant baculoviruses [9].
- High-Five (BTI-Tn-5B1-4): This cell line originated from a different insect species, the cabbage looper Trichoplusia ni [12]. It was developed from embryonic tissue by the Boyce Thompson Institute (BTI) to offer an alternative with often superior recombinant protein expression levels, particularly for secreted proteins [12] [10].
The following diagram illustrates the phylogenetic relationship and key developmental milestones of these cell lines.
Comparative Characteristics
A clear understanding of the distinct properties of each cell line is crucial for selecting the most appropriate one for a given application. The table below summarizes their key characteristics.
Table 1: Comparative Characteristics of Sf9, Sf21, and High-Five Cell Lines
| Feature | Sf21 | Sf9 | High-Five (BTI-Tn-5B1-4) |
|---|---|---|---|
| Species of Origin | Spodoptera frugiperda [7] | Spodoptera frugiperda [11] | Trichoplusia ni [12] |
| Tissue of Origin | Pupal ovarian tissue [7] | Pupal ovarian tissue (clonal isolate of Sf21) [11] | Embryonic tissue [9] [12] |
| Morphology | Spherical, disparate size [9] [7] | Spherical, regular and smaller size [9] [11] | Can be cultured in loose attached state or suspension [12] |
| Doubling Time | ~24 hours [7] | 48-72 hours [7] | Information not in search results |
| Growth in Suspension | Suitable [7] | Excellent; highly tolerant to high densities and shear stress [10] | Excellent; can be cultured in suspension [12] |
| Relative Protein Yield | Suitable for protein expression [10] | High [9] | Very high; often higher than Sf9 for secreted proteins [12] [10] |
| Key Applications | Plaque assays, virus amplification [10] | Virus amplification, high-titer stock production, recombinant protein expression [9] [10] | High-level recombinant protein production, VLP vaccines [12] [13] |
| Notable Features | High susceptibility to baculovirus, forms irregular plaques [9] [10] | Forms uniform monolayers and plaques, moderate virus susceptibility [9] [10] | Can produce abundant small silencing RNAs, serum-free adaptation [12] |
Detailed Subculturing Protocols
Maintaining healthy cell cultures requires adherence to specific protocols. The following methods are effective for the subculturing of adherent and loosely attached insect cell lines [5].
General Preparation
This initial preparation is consistent for all cell line types.
- Step 1: Turn on the laminar flow hood and disinfect the working surface with 70% ethanol.
- Step 2: Examine the mature cell culture under an inverted phase-contrast microscope. The medium should be clear, and the cells should appear refractive. Cloudiness may indicate bacterial contamination.
- Step 3: Record all passage information (date, cell line, passage number, split ratio) in a laboratory record book.
- Step 4: Label new tissue culture flasks with the cell line designation, passage number, and date [5].
Protocol for Loosely Attached & Non-Attached Cells
This method is suitable for cell lines like Trichoplusia ni TN-368 and IAL-TND1. High-Five cells, which can be loosely attached, may also be subcultured using this method or the one below [12] [5].
- Aspirate Medium: Place the bottle of fresh medium, the mature culture, and the new labeled flask(s) in the hood. Using a sterile pipette, transfer the appropriate volume of fresh medium into the new flask. For a 1:10 split in a 25 cm² flask, add 3.6 mL of fresh medium.
- Resuspend and Transfer: Gently swirl the mature culture to evenly disperse the cells. Using a new sterile pipette, draw the required volume of cell suspension (e.g., 0.4 mL for a 1:10 split) and dispense it into the new flask containing fresh medium.
- Incubate: Tighten all caps and place the new culture in a 26–28°C incubator [5].
Protocol for Strongly Adherent Cells
This procedure is optimized for strongly attached cell lines such as Sf9, Sf21, and High-Five [5].
- Chill Culture: Place the mature culture at 4°C for approximately 20 minutes. This step helps in detaching the cells.
- Detach Cells: Remove the culture from the refrigerator and place it in the hood. Strike the flask sharply with the palm of your hand 2-3 times to mechanically dislodge the adherent cells from the surface.
- Add Fresh Medium: Add the calculated volume of fresh medium to the new culture flask (e.g., 3.6 mL).
- Inoculate New Culture: Using a sterile pipette, draw the appropriate volume of the resuspended cell solution from the mature culture (e.g., 0.4 mL) and transfer it to the new flask.
- Incubate: Tighten the caps and place the new culture in a 26–28°C incubator [5].
The workflow for subculturing adherent cells is summarized in the following diagram.
Essential Reagents and Materials
Successful cell culture maintenance depends on the use of specific, high-quality reagents.
Table 2: Research Reagent Solutions for Insect Cell Culture
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Culture Medium | Provides nutrients for cell growth and maintenance. | SF900 II serum-free medium is commonly used; media may be supplemented with 10% FBS [7]. |
| Fetal Bovine Serum (FBS) | Supplement for culture medium; provides growth factors and adhesion factors. | Often used at 10% concentration, though serum-free adaptation is common [7]. |
| Trypsin Diluent / VMF Trypsin | Enzymatic detachment of strongly adherent cells (not always required for insect cells). | 0.05 mg/ml in divalent cation-free PBS [5]. |
| Cryopreservation Medium | Long-term storage of cell stocks. | Typically 10% DMSO in FBS [7]. |
| Trypan Blue Solution | Viability staining for cell counting. | 0.4% solution [5]. |
Applications in Biopharmaceutical Production
The Sf9, Sf21, and High-Five cell lines have proven their critical value in the production of vaccines and therapeutics.
- Vaccine Production: The BEVS using these cell lines is a well-established platform for producing recombinant protein vaccines and Virus-Like Particles (VLPs). Notable examples include:
- Cervarix: A human papillomavirus (HPV) vaccine produced using the insect cell-BEV system [9] [13].
- NVX-CoV2373 (Novavax): A COVID-19 vaccine based on a recombinant spike protein expressed in Sf9 cells [13].
- Influenza Vaccine (FluBlok): A recombinant hemagglutinin vaccine produced in insect cells [13].
- Therapeutic Proteins: The platform is also used for complex therapeutic proteins. For instance, Provenge (sipuleucel-T), a therapeutic for prostate cancer, involves a recombinant protein component produced in insect cells [13].
- Research Tools: Beyond pharmaceuticals, these cell lines are workhorses in academic and industrial research for expressing "tool proteins" for structural biology, enzymology, and functional studies [9].
Concluding Remarks
The Sf9, Sf21, and High-Five cell lines, each with their unique origins and characteristics, form a powerful trio for biotechnological applications. Sf9 cells offer robustness and regularity for reliable virus production and protein expression; Sf21 cells provide high viral susceptibility ideal for initial virus isolation and plaque assays; and High-Five cells often deliver superior yields of recombinant proteins. Mastering the detailed protocols for their subculturing is fundamental to successful outcomes in both basic research and the commercial production of biopharmaceuticals. As genetic engineering continues to enhance their capabilities, such as humanized glycosylation pathways, the utility of these insect cell lines is poised to expand even further.
The Role of Baculovirus Expression Vector Systems (BEVS) in Protein Production
The Baculovirus Expression Vector System (BEVS) is an established and powerful platform for the production of complex recombinant proteins. Since the first successful expression of human interferon-beta in 1983, BEVS has evolved from a research tool to a mainstream commercial manufacturing platform for viral vaccines, gene therapy vectors, and other biopharmaceuticals [14] [15]. Its relevance is underscored by the approval of multiple human and veterinary products, including the Cervarix human papillomavirus vaccine, Flublok influenza vaccine, and Glybera gene therapy [15]. For research focused on subculturing adherent insect cells, BEVS represents a critical application, leveraging the unique biology of the baculovirus- insect cell interaction to achieve high-yield protein production.
The BEVS Platform: Core Components and Workflow
The BEVS platform relies on two fundamental components: an insect cell line and a recombinant baculovirus. The most commonly used baculovirus is Autographa californica multiple nucleopolyhedrovirus (AcMNPV), a virus with a large, circular double-stranded DNA genome (~134 kbp) that provides ample capacity for inserting large or multiple foreign genes [15] [16].
2.1 Insect Cell Lines The choice of insect cell line is a crucial consideration for protein production. The most frequently used lines are derived from the ovarian tissues of Lepidopteran insects [17].
Table 1: Common Insect Cell Lines for BEVS
| Cell Line | Origin | Key Characteristics | Common Applications |
|---|---|---|---|
| Sf9 | Pupal ovarian tissue of Spodoptera frugiperda [17] | High growth rate, tolerant to high densities and shear stress, uniform morphology [17] | Virus amplification, protein synthesis, general protein production [17] |
| Sf21 | Pupal ovarian tissue of Spodoptera frugiperda [17] | Highly susceptible to viral infection, forms clear plaques [17] | Plaque assays, virus titer determination [15] |
| High Five (Tn5) | Ovarian tissue of Trichoplusia ni [15] [16] | Associated with higher recombinant protein expression, particularly for secreted proteins [16] [18] | Production of secreted proteins and antibodies [18] |
2.2 Baculovirus Biology and Engineering Baculoviruses have a biphasic life cycle, producing two virion phenotypes: the Occlusion-Derived Virus (ODV) and the Budded Virus (BV). The BV form, which acquires its envelope from the plasma membrane, is used for infecting cells in culture and is the form harnessed by BEVS [16] [17]. A key innovation was the discovery that the strong viral very late promoters, such as those driving the polyhedrin (polh) and p10 genes, are dispensable for viral replication in cell culture. These promoters can be replaced with a gene of interest (GOI), allowing the virus to reprogram the infected cell to produce the recombinant protein at very high levels [15] [14].
2.3 Workflow for Recombinant Protein Production The standard workflow for producing a recombinant protein using BEVS involves a series of defined steps, from cloning to purification.
Figure 1: BEVS Workflow for Recombinant Protein Production. The process begins with cloning the gene of interest and culminates in the infection of cultured insect cells and protein harvest [15].
The initial step involves cloning the GOI into a transfer plasmid behind a strong baculovirus promoter [15]. This plasmid is then combined with a modified baculovirus genome (e.g., linearized AcMNPV DNA or a bacmid) and introduced into insect cells. Through homologous recombination or transposition, the GOI is inserted into the baculovirus genome, generating a recombinant baculovirus [15] [16]. A critical best practice is to use a purified, single plaque to generate a high-titer Working Virus Bank (WVB), as this ensures genetic homogeneity and reproducible infections [15]. This WVB is then used to infect adherent insect cell cultures at the appropriate cell density and confluency for protein production, typically harvesting 48 to 96 hours post-infection [15].
Applications in Biomedical Research and Drug Development
BEVS is particularly well-suited for producing complex proteins that are difficult to express in prokaryotic systems. Its applications are extensive and growing.
Table 2: Key Applications of BEVS in Biomedicine
| Application Area | Description | Examples |
|---|---|---|
| Vaccine Development | Production of subunit vaccines and Virus-Like Particles (VLPs) that mimic native virus structures without being infectious. | Cervarix (HPV VLP vaccine) [15]NVX-CoV2373 (Novavax COVID-19 vaccine) [19]Respiratory Syncytial Virus (RSV) F nanoparticle vaccine [19] |
| Gene Therapy Vectors | Manufacturing of recombinant adeno-associated virus (rAAV) vectors used to deliver therapeutic genes. | Glybera (for lipoprotein lipase deficiency) [15] |
| Complex Therapeutic Proteins | Expression of structurally intricate proteins requiring eukaryotic post-translational modifications. | Provenge (Sipuleucel-T) cancer immunotherapy [15]Monoclonal antibodies and Fc-fusion proteins [18] [19] |
| Veterinary Vaccines | Commercial production of safe and effective vaccines for animals. | Porcilis Pesti (classical swine fever) [15]Ingelvac CircoFLEX (porcine circovirus) [15] |
BEVS in Practice: Protocols for the Research Scientist
This section provides actionable protocols for researchers working with adherent insect cell cultures and BEVS.
4.1 Protocol: Subculturing Adherent Sf9 and Sf21 Cells Maintaining healthy, log-phase insect cells is fundamental to successful BEVS protein production.
Materials:
- Healthy, sub-confluent monolayer of Sf9 or Sf21 cells
- Complete insect cell medium (e.g., SF-900 II, EX-CELL 405)
- Sterile 1X PBS (without Ca2+/Mg2+)
- Trypsin-EDTA solution (e.g., 0.25%)
- T-flasks or other culture vessels
- Water bath (27°C)
- Hemocytometer or automated cell counter
Procedure:
- Preparation: Pre-warm all media and reagents to 27°C. Aseptically remove and discard the spent medium from the culture flask.
- Rinsing: Gently rinse the cell monolayer with sterile 1X PBS to remove residual serum and calcium that can inhibit trypsin.
- Trypsinization: Add enough trypsin-EDTA solution to cover the monolayer (e.g., 2-3 mL for a T-75 flask). Incubate at 27°C for 5-10 minutes. Observe cells under a microscope until they round up and detach.
- Neutralization: Add a sufficient volume of complete medium (containing serum or other trypsin inhibitors) to neutralize the trypsin. Typically, use a 2:1 to 4:1 ratio of medium to trypsin.
- Seeding: Perform a cell count and seed new culture flasks at a density of 3.0 x 10^6 cells per T-75 flask in 15-20 mL of fresh, pre-warmed medium [17]. Maintain cultures at 27°C without CO₂.
- Schedule: A subcultivation ratio of 1:3 to 1:4 is generally recommended every 3-4 days to keep cells in their logarithmic growth phase, which is critical for high-efficiency viral infection [17].
4.2 Protocol: Titration of Baculovirus Stock by Plaque Assay Determining the precise titer of your WVB is essential for standardizing infections (Multiplicity of Infection, or MOI) and ensuring reproducible protein yields.
Materials:
- Sf21 cells (recommended for clear plaque formation) [17]
- Low-melting-point agarose
- Baculovirus stock for titration
- Complete growth medium
- Sterile 1X PBS
- 6-well or 12-well tissue culture plates
Procedure:
- Seed Cells: Seed Sf21 cells in a multi-well plate to achieve 50-70% confluency and allow them to attach.
- Prepare Dilutions: Serially dilute the baculovirus stock (e.g., 10^-2 to 10^-8) in sterile medium.
- Infect: Remove medium from the wells and carefully overlay each well with a different dilution of the virus. Incubate for 1 hour at 27°C with gentle rocking every 15 minutes.
- Overlay with Agarose: Prepare a mixture of equal parts 2% low-melting-point agarose and 2X culture medium. After the infection period, remove the viral inoculum and carefully overlay the cell monolayer with the agarose-medium mixture. Allow it to solidify at room temperature.
- Incubate and Count: Add a small amount of medium on top of the agarose to prevent drying. Incubate the plate at 27°C for 5-7 days. Score plaques (clear zones of cell lysis) and calculate the viral titer in Plaque-Forming Units per mL (PFU/mL) using the formula: Titer (PFU/mL) = (Number of plaques) / (Dilution factor x Infection volume (mL)).
- Optimal MOI: For protein production, an MOI of 0.1 to 5 is often used to balance high infection rates with the preservation of cell viability for sufficient protein production time.
4.3 The Scientist's Toolkit: Key Reagents for BEVS Table 3: Essential Research Reagents for BEVS Experiments
| Reagent / Solution | Function | Example Products / Notes |
|---|---|---|
| Transfer Vectors | Plasmids for inserting the GOI into the baculovirus genome. | pFastBac, pOET, pTriEx (allows expression in insect, bacterial, and mammalian cells) [16] [20] |
| Baculovirus DNA / Bacmid | Engineered viral genome for recombination. | BacPAK6, BaculoGold, flashBAC, Bac-to-Bac bacmid [16] [20] |
| Insect Cell Culture Medium | Provides nutrients for cell growth and maintenance. | SF-900 II/III, EX-CELL 405, Grace's Insect Medium [17] [18] |
| Cell Dissociation Reagent | Detaches adherent cells for subculturing and seeding. | Trypsin-EDTA (0.25%) [17] |
| Transfection Reagent | Facilitates delivery of DNA into insect cells for virus generation. | Polyethylenimine (PEI), Cationic lipid-based reagents [18] |
| Agarose (LMP) | Used for plaque assay overlays to isolate viral plaques. | Low-melting-point (LMP) agarose for easy overlaying [17] |
Advanced Strategies and Recent Innovations
The BEVS platform continues to evolve with engineering strategies designed to overcome limitations and enhance protein yield and quality.
Figure 2: BEVS Optimization Strategies. Research efforts focus on addressing key limitations through targeted genetic engineering of the virus, the host cell, or the expression method itself [21] [17] [18].
Recent research demonstrates that overexpression of the essential baculovirus transactivators IE0 and IE1 can significantly boost both viral titers and recombinant protein expression, providing a novel vector optimization strategy [21]. Furthermore, the development of baculovirus-free insect cell expression systems is gaining traction. By using plasmid transfection instead of viral infection, this approach maintains host cell secretory pathway integrity, resulting in higher yields and quality of secreted proteins like antibodies [18] [19].
The Baculovirus Expression Vector System remains a versatile and robust platform for producing a wide array of complex biologics. Its relevance to research involving the subculturing of adherent insect cells is paramount, as the health and status of the host cell culture directly dictate the success of protein production runs. Mastery of core techniques—such as cell culture maintenance, virus titration, and infection—combined with an understanding of emerging optimization strategies, empowers researchers and drug developers to fully leverage the potential of BEVS for advancing therapeutics and biomedical research.
Insect cell lines have become a cornerstone of modern biopharmaceutical manufacturing, offering a unique balance of eukaryotic processing capabilities and prokaryotic economic efficiency. Within the specific context of research on subculturing adherent insect cells, these advantages are not merely theoretical but translate into tangible benefits for the production of recombinant proteins, vaccines, and other complex biologics [14]. For scientists and drug development professionals, understanding how to leverage these properties through optimized protocols is crucial for improving yield, reducing costs, and accelerating time-to-market for therapeutic products.
This document details the practical application of adherent insect cell culture, framing its core advantages—cost-effectiveness, scalability, and comprehensive post-translational modifications—within the framework of hands-on laboratory procedures. We provide summarized quantitative data, detailed experimental protocols, and visual workflows to serve as a comprehensive guide for researchers aiming to harness the full potential of this powerful expression platform.
Core Advantages: A Quantitative and Qualitative Analysis
The adoption of insect cell systems, particularly for adherent culture, is driven by three interconnected pillars of superiority. The tables below summarize key quantitative and qualitative data for easy comparison.
Table 1: Quantitative Advantages of Insect Cell Expression Systems
| Advantage | Quantitative Metric | Comparison/Context |
|---|---|---|
| Protein Yield | Expression levels up to 500 mg/L have been reported [22] [23] | Demonstrates high productivity suitable for industrial-scale manufacturing. |
| Economic Operational Cost | Incubation at 27°C ± 1°C; no CO2 requirement [22] [1] | Significant energy savings compared to mammalian systems (37°C, 5% CO2). |
| Scalability | Successful scale-up to 5L Erlenmeyer flasks at 90% fill volume [24] | Enables cost-efficient R&D scale-up with standard laboratory equipment. |
| Glycosylation Capability | Capable of N-glycosylation, phosphorylation, and acetylation [25] | Produces proteins more similar to native mammalian proteins than yeast or bacterial systems. |
Table 2: Qualitative Advantages and Considerations for Insect Cell Lines
| Aspect | Advantage | Consideration/Limitation |
|---|---|---|
| Cost-Effectiveness | Lower biosafety requirements (BSL1); reduced media and energy costs [26] [25] | Initial setup requires optimization of serum-free media for adherent cultures. |
| Scalability & Culture | Adaptable to high-density suspension and adherent culture; robust growth in simple media [23] [25] | Adherent cells can attach very tightly, requiring careful passaging [1]. |
| Post-Translational Modifications (PTMs) | Performs complex PTMs approaching those of mammalian cells [22] [25] | Glycosylation patterns are not identical to human ones (e.g., lack of sialic acid), which can be addressed via cell line engineering [26] [17]. |
| Expression System | Baculovirus Expression Vector System (BEVS) allows for high-yield expression and co-expression of multiple subunits [26] [23] | BEVS leads to cell lysis, complicating purification; viral contamination is a concern [26]. |
Detailed Experimental Protocols
Protocol 1: Subculturing Adherent Insect Cells
Principle: Maintaining healthy, log-phase adherent insect cell cultures requires regular passaging before confluency to ensure optimal growth and productivity. This protocol is specifically adapted for Sf9, Sf21, and High Five cells [1] [17].
Materials:
- Cell Line: Sf9, Sf21, or High Five cells [17].
- Growth Medium: Sf-900 II SFM or Express Five SFM, pre-warmed [22].
- Dissociation Reagent: Trypsin or TrypLE alternative.
- Balanced Salt Solution: Without calcium and magnesium.
- Equipment: T-flasks, centrifuge, hemocytometer or automated cell counter.
Procedure:
- Assessment: Observe cells under a microscope. Passage when cells are in the log phase and before they reach confluency. Do not allow cells to become over-confluent, as this decreases doubling times and viability [1].
- Medium Removal: Aspirate and discard the spent culture medium completely.
- Washing: Gently add a balanced salt solution without calcium and magnesium to the side of the flask (approx. 2 mL per 10 cm² surface area). Rock the vessel back and forth, then remove and discard the wash solution. This step removes serum and ions that inhibit dissociation [1].
- Cell Detachment: Add pre-warmed dissociation reagent to cover the cell layer (approx. 0.5 mL per 10 cm²). Rock the flask for complete coverage.
- Incubation: Incubate the flask at room temperature for approximately 2 minutes. Actual time may vary; observe cells under a microscope. For strongly adherent insect cells under serum-free conditions, a quick, sharp shake of the flask may be necessary. Avoid vigorous shaking to prevent cell damage [1].
- Neutralization: When ≥90% of cells are detached, add twice the volume of the dissociation reagent of pre-warmed complete growth medium. Pipette the medium over the cell layer surface several times to ensure a single-cell suspension.
- Centrifugation: Transfer the cell suspension to a conical tube and centrifuge at 200 × g for 5–10 minutes.
- Resuspension and Seeding: Discard the supernatant. Resuspend the cell pellet in a small volume of fresh, pre-warmed medium. Perform a cell count and viability assessment (e.g., Trypan blue exclusion). Dilute the cell suspension to the recommended seeding density and pipette into new culture vessels.
- Incubation: Loosen the caps on the new flasks and place them in a 27°C incubator. No CO₂ control or humidity is required. Protect cultures from light [22] [1].
Protocol 2: Recombinant Protein Production via BEVS
Principle: The Baculovirus Expression Vector System (BEVS) uses a recombinant baculovirus to infect adherent insect cells, leveraging strong viral promoters to drive high-level protein expression 48–72 hours post-infection [26] [22].
Materials:
- Cell Line: Sf9 or Sf21 cells (highly susceptible to baculovirus infection) [17].
- Baculovirus System: e.g., Bac-to-Bac or BaculoDirect kit [22].
- Virus Amplification & Titering: Low-melting-point agarose, 6-well plates.
- Infection: High-titer virus stock (≥1 x 10⁸ PFU/mL recommended) [22].
Procedure:
- Virus Generation (Bac-to-Bac Example): Clone the GOI into a pFastBac donor plasmid. Transform this into DH10Bac E. coli cells to generate a recombinant bacmid via transposition. Isolate the bacmid DNA [26] [17].
- P1 Virus Production: Transfect the recombinant bacmid DNA into adherent Sf9 cells using a transfection reagent. Harvest the supernatant (P1 virus stock) 5–7 days post-transfection.
- Virus Amplification & Titering: Infect fresh adherent Sf9 cells with a small volume of P1 stock to generate a P2 stock. Amplify further if needed. Determine the viral titer using a plaque assay [22]:
- Plate Sf21 cells (ideal for clear plaques) at 80% confluence in a 6-well plate.
- Infect with serial dilutions of the virus stock.
- Overlay with medium containing 1% low-melting-point agarose.
- Incubate for 10–14 days, then count plaques. Calculate titer as PFU/mL.
- Protein Expression: Seed adherent insect cells to reach ~70-80% confluency at the time of infection. Infect cells at a Multiplicity of Infection (MOI) of 5–10 with the high-titer virus stock [22].
- Harvesting: Express the protein for 48–72 hours. Do not exceed 72 hours, as cell lysis and protease release can degrade the product. Harvest the supernatant (for secreted proteins) or lyse the cells (for intracellular proteins) for subsequent purification.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Materials for Insect Cell Culture and Protein Expression
| Item | Function/Application | Example Products / Notes |
|---|---|---|
| Sf-900 II/III SFM | Serum-free medium (SFM) optimized for growth of Sf9 and Sf21 cells in suspension and adherent culture. | Gibco Sf-900 II SFM; supports high-density growth without shear-force protectants [22]. |
| Express Five SFM | Serum-free medium formulated for the culture and protein expression of High Five cells. | Requires supplementation with glutamine [22]. |
| Bac-to-Bac System | A rapid and efficient baculovirus expression system that generates recombinant bacmids in E. coli. | Utilizes site-specific transposition; allows for blue/white selection of colonies [26] [22]. |
| BaculoDirect System | A Gateway recombination-based system for rapid generation of recombinant baculovirus. | Ideal for high-throughput; generates purified virus in less than one week [22]. |
| Grace's Insect Medium | A classic complete medium for insect cell culture, can be supplemented with FBS. | Often used with 10% heat-inactivated FBS for certain applications [22]. |
| Pluronic F-68 | A non-ionic surfactant used to protect cells from shear forces in suspension culture. | Typically added to serum-containing media for suspension cultures [22]. |
Insect cell lines present a compelling platform for biotherapeutic development, uniquely combining operational cost-effectiveness, straightforward scalability in both adherent and suspension formats, and eukaryotic post-translational modification capabilities. The protocols and data outlined herein provide a foundation for researchers to reliably subculture adherent insect cells and implement the BEVS for high-yield protein production. Continuous advancements in genetic engineering, such as the development of glycan-humanized cell lines (e.g., SfSWT-1) and anti-apoptotic engineering, are further overcoming historical limitations like non-human glycosylation and virus-induced cell lysis [26] [17] [14]. By integrating these optimized application notes, scientists and drug development professionals can more effectively leverage this powerful technology to accelerate the discovery and manufacturing of next-generation biologics.
Within the biopharmaceutical industry, the baculovirus/insect cell expression system is a versatile platform for producing complex biologics, including recombinant proteins and virus-like particles (VLPs) [27]. Adherent insect cell cultures are central to this system, requiring specific environmental conditions distinct from mammalian cell culture. Unlike mammalian cells, which are typically maintained at 37°C in a humidified, CO2-enriched atmosphere, insect cells such as Sf9 and Hi5 require incubation at 27°C in a non-humidified environment [1] [28]. This protocol details the essential laboratory setup and methods for the subculture of adherent insect cells, providing a foundational framework for research and development in drug discovery and bioproduction.
Key Environmental Parameters for Insect Cells
The physiological requirements of insect cells differ significantly from those of mammalian cells. Adherence to these parameters is crucial for maintaining cell viability, growth, and productivity.
Table 1: Key Environmental Parameters for Adherent Insect Cell Culture
| Parameter | Optimal Condition | Notes |
|---|---|---|
| Incubation Temperature | 27°C | A controlled 27°C environment is recommended, though cells can be maintained at room temperature on the bench top if protected from light [1] [28]. |
| Humidity | Non-humidified | Unlike mammalian culture, a humidified atmosphere is not required for insect cells [1] [28]. |
| CO₂ | Not required / Not recommended | CO₂ exchange is not recommended for insect cell culture [1] [28]. |
| Culture Surface | Growth-promoting substrate | Insect cells are anchorage-dependent and require attachment to a surface for proliferation [1]. |
Materials and Reagent Solutions
Table 2: Research Reagent Solutions for Insect Cell Culture
| Item | Function | Example / Notes |
|---|---|---|
| Cell Line | Host for protein/VLP production | Sf9 (ATCC-CRL-1711), BTI-TN-5B1-4 (Hi5), Tnms42 (TN42) [29] [27]. |
| Growth Medium | Provides nutrients and pH balance | Grace's Insect Medium, SFM4 Insect Medium [27]. Formulations are often more acidic than mammalian media [1]. |
| Dissociation Reagent | Detaches adherent cells for subculturing | Enzymatic (e.g., trypsin) or chemical agents. Strongly adherent cells may require mechanical force [1]. |
| Surfactant | Protects cells from shear stress | 0.1% Pluronic F-68 is recommended for suspension adaptation; some media include surfactants [28]. |
| Cryoprotectant | Prevents ice crystal formation during freezing | DMSO (5–10%) is commonly used [4]. |
Protocols for Subculturing Adherent Insect Cells
Routine Passaging of Adherent Insect Cells
This protocol describes the dissociation and subculturing of adherent insect cells.
Step 1: Monitoring and Assessment
- Routinely monitor cells and passage at the log phase of growth. For strongly adherent cells, passage may occur at confluency or slightly after when cells begin to pull away from the flask [1].
- Note: Repeatedly passaging cells either too early (before confluency) or too late (past confluency) can result in decreased doubling times, decreased viability, and poor health [1].
Step 2: Washing
- Remove and discard the spent cell culture media.
- Wash the cell layer using a balanced salt solution without calcium and magnesium to remove traces of serum that would inhibit trypsin [1].
Step 3: Dissociation
- Add a pre-warmed dissociation reagent (e.g., trypsin) to cover the cell layer.
- Incubate the vessel at room temperature for approximately 2 minutes (actual time varies by cell line).
- Observe under a microscope. For strongly adherent insect cells, you may need to tap the vessel or give it one quick, wrist-snapping shake (tighten the cap first) to dislodge cells. Do not shake vigorously [1].
Step 4: Neutralization and Collection
- When ≥90% of cells are detached, add pre-warmed complete growth medium (twice the volume of dissociation reagent used) to neutralize the enzyme.
- Transfer the cell suspension to a centrifuge tube and pellet cells at ~200 x g for 5-10 minutes [1].
Step 5: Seeding New Cultures
Cryopreservation of Insect Cells
Preserving cells at early passages maintains genetic stability and prevents contamination [4].
Step 1: Harvesting
- Harvest a healthy, log-phase culture as described in the passaging protocol (Steps 1-4).
- After centrifugation, discard the supernatant.
Step 2: Resuspension in Cryoprotectant
- Gently resuspend the cell pellet in an appropriate cryoprotectant, such as 5-10% DMSO in serum or serum-containing media, at a density of 1–2x10^6 cells per mL for adherent cells [4].
- Transfer the suspension to labeled cryovials.
Step 3: Controlled Freezing and Storage
- Place cryovials in an isopropanol-based (e.g., Mr Frosty) or alcohol-free polyethylene (e.g., CoolCell) freezing container.
- Store at -80°C overnight to allow a controlled freezing rate of approximately -1°C per minute.
- The next day, transfer vials to the vapor phase of liquid nitrogen for long-term storage [4].
The following workflow diagram illustrates the logical relationship between the different protocol stages and their key decision points.
Experimental Data and Optimization
Shear Stress Resistance in Suspension
While this article focuses on adherent culture, adapting cells to suspension is often necessary for scale-up. Recent research demonstrates that insect cells exhibit high shear resistance.
Table 3: Shear Stress Experimental Data on Insect Cells
| Cell Line | Experimental Setup | Key Finding | Implication |
|---|---|---|---|
| Sf9 & Hi5 | Microfluidic shear device generating defined shear rates [29]. | Cells displayed high resistance to shear rates up to 8.73 × 10⁵ s⁻¹ [29]. | Challenges the historical hypothesis of high insect cell shear sensitivity. |
| Sf9 & Hi5 | Cultivation in microbial Continuous Stirred-Tank Reactors (CSTRs) [29]. | No negative impact on cell viability at high revolution speeds and low aeration rates [29]. | Enables process redesign with high stirring speeds (improving oxygen transfer) and low aeration (reducing foam and bubble damage). |
Optimizing Suspension Culture Parameters
A study optimizing HEK293 cells for suspension culture highlights parameters relevant to insect cell bioprocess development. The optimal conditions identified were an agitation rate of 110 RPM, an orbital diameter of 25 mm, and a relative humidity of 85%, yielding the highest specific growth rate and shortest doubling time [30]. This underscores the importance of systematic parameter optimization.
Discussion
The specific requirement for a 27°C and non-humidified environment is a fundamental aspect of insect cell physiology that directly impacts laboratory setup and operational protocols. Adherence to these conditions, combined with precise subculturing techniques, ensures robust cell growth and reliable experimental outcomes.
The revelation that insect cells possess high shear resistance [29] provides a scientific basis for reevaluating traditional process designs. This knowledge allows researchers to develop more robust and scalable cultivation strategies, potentially leveraging microbial-grade bioreactors to overcome limitations like oxygen transfer and foaming.
Future directions in insect cell culture include advanced genetic engineering to improve productivity and control, such as CRISPR/Cas9 systems to reduce contaminating baculovirus in VLP production runs [27]. As the demand for complex biologics and viral vectors grows, optimized and scalable insect cell culture protocols will remain indispensable in biopharmaceutical research and development.
Step-by-Step Protocol: Passaging and Maintaining Healthy Adherent Insect Cultures
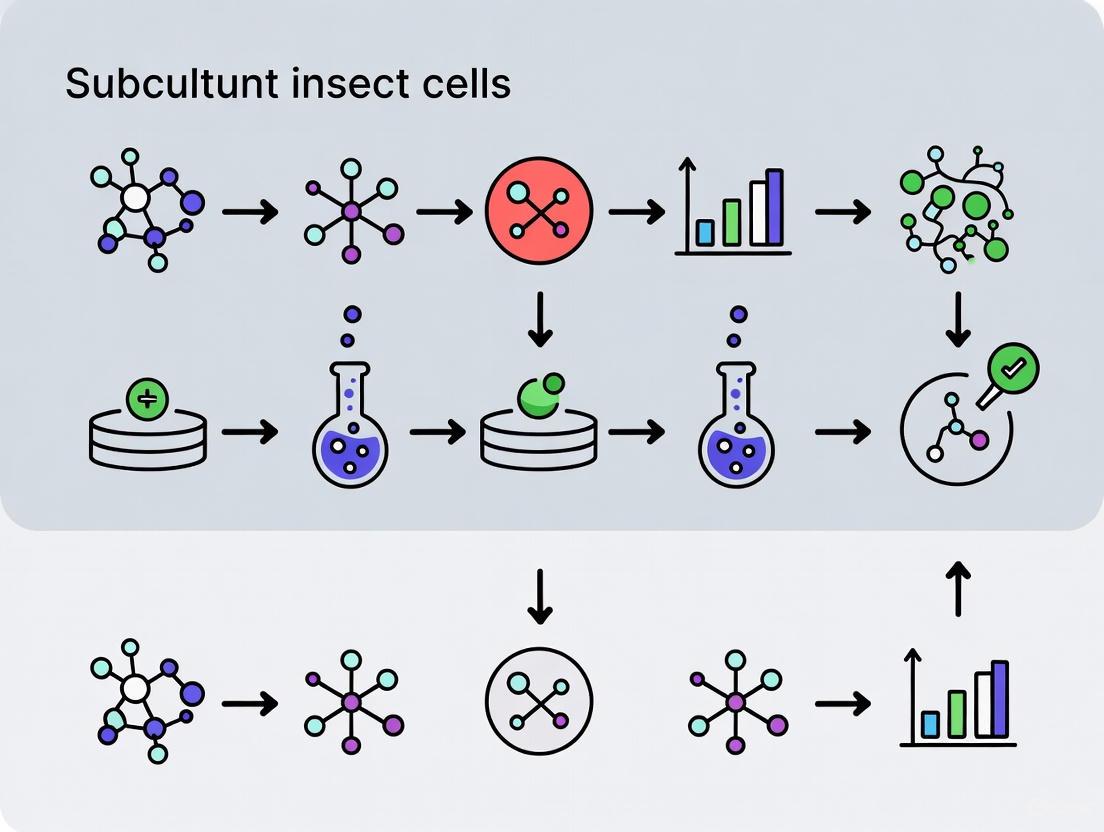
The successful subculturing of adherent insect cells is a cornerstone of modern biopharmaceutical production, enabling critical applications in vaccine development, recombinant protein production, and gene therapy research [14] [31]. Unlike their mammalian counterparts, adherent insect cells present unique challenges in detachment due to their strong attachment to substrates, particularly under serum-free conditions [1]. The selection of appropriate culture media and dissociation agents is therefore not merely a procedural step but a critical determinant of cell viability, functionality, and experimental reproducibility. This application note provides a detailed framework for selecting and utilizing these essential reagents, framed within the broader context of optimizing subculturing protocols for adherent insect cell lines. By integrating current market data with established and emerging protocols, this guide serves to empower researchers and drug development professionals in making informed decisions that enhance yield, maintain cell health, and ensure the consistency required for rigorous scientific and manufacturing standards.
The Research Reagent Toolkit
A successful subculture of adherent insect cells requires a carefully selected suite of reagents. Each component plays a specific role in maintaining cell health during the passaging process. The table below catalogs the essential materials and their functions.
Table 1: Essential Research Reagents for Subculturing Adherent Insect Cells
| Reagent Category | Specific Examples | Primary Function |
|---|---|---|
| Growth Media | Grace's Insect Medium, Serum-Free Formulations, Chemically Defined Media [1] [31] | Provides nutrients, vitamins, salts, and energy sources to support cell growth and proliferation. |
| Dissociation Agents | Trypsin, TrypLE, Accutase, Enzyme-Free Cell Dissociation Buffers [32] | Enzymatically or chemically disrupts cell attachments to the growth surface. |
| Balanced Salt Solution (BSS) | Calcium- and Magnesium-Free PBS, EDTA solutions [1] [32] | Washes away residual serum and ions that inhibit dissociation agents; used in cell rinsing. |
| Culture Vessels | T-flasks, Roller Bottles, Multi-tray Systems [33] | Provides a sterile, growth-promoting substrate for cell attachment and expansion. |
| Cryoprotectants | DMSO, Glycerol, Commercial Preparations (e.g., Bambanker) [4] | Protects cells from ice crystal formation and osmotic shock during freezing. |
Media Selection and Market Landscape
The foundation of healthy insect cell culture is the growth medium. As of 2025, the global insect cell culture media market is valued at approximately $250 million, with a projected compound annual growth rate (CAGR) of 8% through 2033 [34]. This growth is propelled by rising demand for biopharmaceuticals and advancements in cell-based therapies.
Media Formulations and Applications
Insect cells are typically cultured in media that are more acidic than those used for mammalian cells, with Grace's medium being a common historical example [1]. The current market offers two primary physical forms of media: liquid and powder. Liquid media dominates the market due to its convenience and immediate applicability, reducing preparation time and potential for error [34]. Powdered media, while offering a longer shelf life and cost-effectiveness for large-scale operations, shows steady growth in industrial settings [34].
A significant trend is the shift towards serum-free and chemically defined media [34] [31]. These formulations minimize batch-to-batch variability, improve reproducibility, reduce contamination risks, and enhance regulatory compliance—a critical consideration for biopharmaceutical manufacturing. Furthermore, vendors are increasingly tailoring media formulations to meet the unique nutritional needs of specific insect cell lines (e.g., Sf9, Sf21, High Five) to optimize growth and protein production [35] [34].
Table 2: Insect Cell Culture Media: Quantitative Market and Application Analysis
| Segment | 2025 Market Valuation & Growth | Key Characteristics & Applications |
|---|---|---|
| Global Market | ~$250 Million, 8% CAGR (2025-2033) [34] | Driven by biopharmaceutical demand, vaccine production, and recombinant protein needs. |
| Media Type | Liquid Media: Dominates market share [34]Powder Media: Steady growth for large-scale use [34] | Liquid: Convenience, ease-of-use.Powder: Cost-effective, long shelf-life. |
| Formulation | Serum-Free & Chemically Defined: Key growth trend [34] [31] | Enhances reproducibility, reduces variability, and supports regulatory compliance. |
| Application | Scientific Research: Largest application segment [34]Industrial: Rapid growth in biomanufacturing [34] | Research: Protein expression, fundamental studies.Industrial: Vaccine, therapeutic protein production. |
The following diagram illustrates the logical decision-making process for selecting the appropriate culture media based on project goals and requirements.
Dissociation Agent Selection and Protocols
Detaching adherent insect cells requires careful selection of dissociation agents and strict adherence to protocol. Insect cells can attach very tightly, sometimes necessitating mechanical force in addition to chemical agents [1].
Types of Dissociation Agents
A variety of agents can be used to disrupt the extracellular matrix and cell-surface proteins. The choice depends on the cell line's adherence strength and sensitivity.
Table 3: Comparison of Cell Dissociation Agents and Techniques
| Dissociation Agent/Technique | Mechanism of Action | Typical Applications & Considerations |
|---|---|---|
| Trypsin [32] | Proteolytic enzyme that digests cell-adhesion proteins. | Effective for strongly adherent cells; requires precise incubation time to avoid damage. |
| TrypLE [32] | A recombinant fungal protease alternative to trypsin. | A strong, animal origin-free option for robustly adherent cells. |
| Accutase [32] | A blend of proteolytic and collagenolytic enzymes. | A gentler alternative suitable for sensitive cells, including stem cells and primary cells. |
| Enzyme-Free Buffers [32] | Chelating agents (e.g., EDTA) that bind cations required for cell adhesion. | A gentle method that maintains cellular surface proteins; ideal for flow cytometry. |
| Mechanical Dislodgement [1] | Physical force (e.g., a quick, sharp shake) to detach cells. | Often required for tightly adherent insect cells under serum-free conditions. Avoid vigorous shaking. |
Detailed Subculturing Protocol for Adherent Insect Cells
The following step-by-step protocol, adapted from established methodologies, ensures high cell viability and recovery [1] [4] [32].
Materials Required:
- Pre-warmed complete growth medium and dissociation agent (selected from Table 3).
- Balanced salt solution (BSS) without calcium and magnesium (e.g., PBS).
- Culture vessel containing adherent insect cells at appropriate confluency.
- Centrifuge tubes, serological pipettes, and a calibrated pipette.
- Inverted microscope and cell counter (e.g., hemocytometer or automated counter).
Procedure:
- Pre-subculture Observation: Confirm cells are healthy and at the correct confluency for passaging. For many insect cells, this can be at confluency or slightly after, as they may be easier to dislodge [1]. Routinely monitor viability, which should be >90% at subculturing [1] [32].
- Remove Spent Medium: Aspirate and discard the spent cell culture media from the vessel.
- Rinse Cell Layer: Gently wash the cell monolayer using 2-5 mL of pre-warmed BSS per 10 cm² of surface area to remove any traces of serum, calcium, and magnesium that inhibit trypsin and other dissociation agents [1] [32]. Aspirate and discard the wash solution.
- Apply Dissociation Agent: Add pre-warmed dissociation reagent (e.g., ~0.5 mL per 10 cm² for trypsin) to the side of the vessel opposite the cell layer to avoid disruption. Gently rock the vessel to ensure complete coverage [1].
- Incubate and Monitor: Incubate the culture vessel at room temperature for approximately 2 minutes. Observe cells under a microscope every 30 seconds for detachment. If less than 90% of cells are detached, increase incubation time incrementally [1]. For stubborn insect cells, a quick, sharp shake with a tightened cap may be necessary [1]. Do not shake vigorously, as this can damage cells.
- Neutralize Reaction: When ≥90% of cells are detached, tilt the vessel to drain the cells. Add 2 volumes of pre-warmed complete growth medium (relative to the volume of dissociation reagent used) to neutralize the enzyme. Disperse the medium by pipetting over the cell layer surface several times to ensure a single-cell suspension [1] [32].
- Transfer and Centrifuge: Transfer the cell suspension to a centrifuge tube and pellet the cells at ~200 × g for 5-10 minutes [1]. Note that centrifuge speed and time may vary based on cell type.
- Resuspend and Count: Resuspend the cell pellet in a small volume of fresh, pre-warmed complete growth medium. Remove a sample for counting and viability assessment using Trypan blue exclusion or an automated cell counter [1].
- Seed New Cultures: Dilute the cell suspension to the recommended seeding density for the specific insect cell line. Pipette the appropriate volume into new culture vessels and return them to the incubator. Maintain insect cells at 27°C in a non-humidified environment; CO2 exchange is not recommended [1].
The workflow below summarizes the key stages of this subculturing process.
Discussion and Concluding Remarks
The strategic selection of media and dissociation agents is paramount for maintaining robust and reproducible adherent insect cell cultures. The current market trajectory emphasizes a definitive shift towards serum-free, chemically defined media, which enhance process control and regulatory compliance—critical factors for therapeutic and vaccine development [34] [31]. Similarly, the choice of dissociation agent must be tailored to the specific insect cell line, balancing the need for efficient detachment with the preservation of cell surface integrity and viability.
Future advancements in this field will likely be driven by continued cell-line engineering, particularly using tools like CRISPR/Cas9 to modify glycosylation pathways and create more human-like post-translational modifications [14] [31]. Furthermore, the development of stable, baculovirus-free insect cell lines promises to reduce variability and simplify scale-up for manufacturing [31]. By adhering to the detailed protocols and selection criteria outlined in this application note, researchers can effectively navigate the complexities of subculturing adherent insect cells, thereby supporting the advancement of biopharmaceutical research and production.
Within the broader research on subculturing adherent insect cells, the assessment of cell health and the determination of the optimal passaging time are foundational to experimental reproducibility and success. Properly timed subculture is critical for maintaining cells in their logarithmic growth phase, which is essential for robust viability and high yield in downstream applications such as recombinant protein production using the baculovirus expression vector system (BEVS) [10]. This application note details the protocols for evaluating the health of adherent insect cell cultures and establishing the critical passaging window.
Assessing Cell Health: Morphological and Quantitative Indicators
Routine monitoring of cell health is a prerequisite for successful subculturing. Assessment should combine morphological observation with quantitative viability measurements.
Morphological Features of Healthy vs. Unhealthy Cells
Daily observation under an inverted phase contrast microscope is essential. The medium should be relatively clear, and healthy cells should appear refractive and possess a regular, spherical morphology [10] [5]. The table below summarizes key morphological indicators.
Table 1: Morphological Indicators of Cell Health in Adherent Insect Cultures
| Aspect | Healthy Cells | Unhealthy Cells |
|---|---|---|
| General Appearance | Refractive and spherical [5] | Granular, vacuolated, or irregular shape |
| Membrane Integrity | Smooth, intact membrane | Blebbing or broken membrane |
| Culture Medium | Clear, not cloudy [5] | Cloudy, which may suggest bacterial contamination [5] |
| Cell Density | Monolayer at optimal confluency | Over-confluent or excessively sparse |
Quantitative Assessment of Viability
Cell viability, a critical quantitative metric, should be assessed at the time of subculturing. Viability should be greater than 90% before passaging [1]. Viability can be determined using methods such as the Trypan blue exclusion assay and counted manually with a hemocytometer or automatically with a cell counter [1]. Trypan blue is a dye that is excluded by live cells with intact membranes but penetrates and stains dead cells [5].
Determining the Optimal Time for Passaging
Passaging at the correct time is crucial for maintaining culture health. For adherent insect cells like Sf9 and Sf21, this means passaging at log phase, before they reach confluency, unless they are strongly adherent [1].
Passaging Guidance for Specific Cell Lines
The optimal confluency for passaging can vary depending on the specific cell line and its adherence properties. The table below provides guidance based on common insect cell lines used in research.
Table 2: Optimal Passaging Parameters for Common Adherent Insect Cell Lines
| Cell Line | Origin | Recommended Passaging Confluency | Key Characteristics for Passaging |
|---|---|---|---|
| Sf21 | Pupal ovarian tissue of Spodoptera frugiperda [10] | Before confluency [1] | Strongly adherent; passages at confluency or slightly after when cells begin to pull away from the flask [1]. |
| Sf9 | Subclone of Sf21 [10] | Before confluency [1] | Spherical, more regular size; highly tolerant to high densities and shear stress [10]. |
| Strongly Adherent Strains (e.g., IPLB-LdEIta, UFL-AG286) | Various insect tissues [5] | At confluency or slightly after [1] | Require passaging when cells start to detach; repeated passaging at high densities decreases health [1]. |
Consequences of Improper Passaging Timing
Deviating from the optimal passaging window has direct consequences for culture health:
- Passaging Too Early (Low Density): Inhibits cell growth. For insect cells, densities lower than 20% confluency inhibit growth [1].
- Passaging Too Late (Post-Confluency): Leads to decreased doubling times, decreased viability, and a diminished ability of cells to attach in subsequent cultures [1].
Experimental Protocol: Workflow for Assessing and Passaging Cells
The following integrated protocol outlines the steps from initial assessment to subculturing.
Detailed Methodology for Passaging Adherent Insect Cells
Materials Required:
- Mature cell culture in late log phase [5]
- Appropriate insect cell growth medium (e.g., Grace's Medium) [1]
- Laminar flow hood [1] [5]
- Inverted phase contrast microscope [5]
- Sterile pipettes and centrifuge tubes [1]
- Refrigerated incubator set to 27°C [1]
Procedure:
- Preparation and Examination: Turn on the laminar flow hood and wipe the surface with 70% ethanol [5]. Examine the mature cell culture under an inverted microscope. The medium should be clear, and cells should be refractive [5]. Record passage information [5].
- Cell Detachment (for strongly adherent cells like Sf9 and Sf21):
- For strongly adherent insect cells that attach very tightly, standard dissociation reagents may be insufficient. To dislodge the cells, you may need to strike the flask sharply on the side with the palm of your hand two or three times [5]. To avoid contamination, always tighten the cap before this procedure. Do not shake the flask vigorously, as it may damage the cells [1].
- Alternatively, as per general adherent cell protocol, wash the cell layer with a balanced salt solution without calcium and magnesium to remove traces of serum. Add a pre-warmed dissociation reagent and incubate at room temperature for approximately 2 minutes, observing under the microscope for detachment [1].
- Neutralization and Cell Collection: When ≥ 90% of cells have detached, add a volume of complete growth medium equivalent to twice the volume of the dissociation reagent used to neutralize the reaction [1]. Transfer the cell suspension to a conical tube.
- Centrifugation and Resuspension: Centrifuge the cells at 200 x g for 5 to 10 minutes [1]. Resuspend the cell pellet in a small volume of fresh, pre-warmed growth medium [1].
- Cell Counting and Viability Assessment: Remove a sample of the cell suspension for counting. Determine the total cell count and percent viability using a hemocytometer and Trypan blue exclusion protocol or an automated cell counter [1]. Only proceed if viability is >90% [1].
- Seeding New Cultures: Dilute the cell suspension with fresh medium to the recommended seeding density for your cell line. Pipette the appropriate volume into new culture vessels. Incubate the new cultures at 27°C in a non-humidified environment [1].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Materials for Adherent Insect Cell Culture
| Item | Function / Application | Example / Note |
|---|---|---|
| Insect Cell Growth Medium | Provides nutrients and environment for cell growth. | Grace's Insect Medium, formulated for insect cells and more acidic than mammalian media [1]. |
| Dissociation Reagent | Detaches adherent cells from the culture vessel surface. | Trypsin or TrypLE; may require mechanical force for strongly adherent insect cells [1] [5]. |
| Cryoprotectant | Protects cells from ice crystal formation during freezing. | DMSO (5-10%) or Glycerol (2-20%); DMSO can be toxic to some cells [4]. |
| Cell Counter / Hemocytometer | Determines cell concentration and viability. | Used with Trypan Blue exclusion method to count live/dead cells [1] [5]. |
| Balanced Salt Solution (without Ca2+/Mg2+) | Washes cell layer before dissociation. | Removes calcium and magnesium that inhibit trypsin activity [1]. |
Within the context of subculturing adherent insect cells for recombinant protein production and vaccine development, the selection of an appropriate cell detachment technique is a critical step that directly impacts cell viability, surface marker integrity, and subsequent experimental reproducibility. Insect cell lines such as Sf9 and Sf21, derived from Spodoptera frugiperda, are widely utilized in baculovirus expression vector systems (BEVS) for their cost-effectiveness and high protein yield capabilities [17]. These cells exhibit particularly strong adherence to culture substrates under serum-free conditions, presenting unique challenges for researchers seeking to maintain cellular health and functionality during subculturing [1]. This application note provides a detailed comparative analysis of mechanical versus enzymatic detachment methods specifically framed within insect cell research, offering evidence-based protocols to guide researchers and drug development professionals in optimizing their subculturing workflows.
Comparative Analysis of Detachment Methods
The fundamental challenge in dissociating strongly adherent insect cells lies in overcoming robust cellular attachments while preserving viability, surface protein integrity, and normal physiological function. The following sections provide a comprehensive comparison of the primary detachment methodologies.
Mechanical Detachment Methods
Mechanical detachment techniques utilize physical force to dislodge cells from culture surfaces and are particularly relevant for strongly adherent insect cell lines.
Shake-off and Scraping: For insect cells that attach very tightly under serum-free conditions, laboratories often employ a "wrist-snapping motion" to dislodge cells, taking care to avoid vigorous shaking that may cause cellular damage [1]. Cell scraping provides an alternative mechanical approach that avoids enzymatic exposure entirely, potentially preserving surface protein integrity [36] [37].
Applications and Limitations: Mechanical methods are optimal when preserving surface protein epitopes is critical for downstream applications such as immunocytochemistry or flow cytometry analysis. However, these techniques may result in inconsistent cell yields and viabilities between users [38] and typically generate heterogeneous cell clusters rather than uniform single-cell suspensions, which can affect subsequent experimental consistency.
Enzymatic Detachment Methods
Enzymatic dissociation employs proteolytic enzymes to digest adhesion proteins that facilitate cell attachment to culture surfaces.
Enzyme Selection: Trypsin remains the most frequently used enzymatic agent for cellular dissociation, effectively cleaving after lysine or arginine residues to degrade most cell surface proteins [36]. For insect cell systems, Accutase is often recommended as a milder-acting enzymatic alternative to trypsin, while TrypLE Express serves as an animal origin-free option suitable for regulatory-sensitive applications [37].
Considerations for Insect Cells: Research demonstrates that enzymatic treatments can compromise specific surface proteins despite their efficiency in cell detachment. Studies show Accutase significantly decreases surface Fas ligands (FasL) and Fas receptors on macrophages, cleaving the extracellular portion of FasL into fragments under 20 kD [36]. This protein degradation requires a recovery period of up to 20 hours for proper surface expression restoration, a critical consideration for time-sensitive experiments [36].
Quantitative Comparison of Detachment Methods
Table 1: Comprehensive comparison of cell detachment techniques for adherent insect cells
| Parameter | Mechanical (Shake-off/Scraping) | Enzymatic (Trypsin/Accutase) | Non-Enzymatic (EDTA-based) |
|---|---|---|---|
| Efficiency on Strongly Adherent Cells | Variable; may require multiple attempts | High; effective for strongly adherent cells | Low to moderate; may require scraping assistance |
| Cell Viability Post-Detachment | Moderate; potential for physical damage | High when optimized (>90%) | High (>90%) |
| Surface Protein Integrity | Preserved | Compromised (e.g., FasL, Fas receptor) | Preserved |
| Recovery Time Required | Minimal | 2-20 hours for protein recovery | Minimal |
| Single Cell Suspension Quality | Poor; clusters common | Excellent | Good |
| Downstream Application Compatibility | Flow cytometry, immunostaining | Subculturing, bulk protein production | Flow cytometry, receptor studies |
| Typical Incubation Time | Immediate | 5-15 minutes | 5-15 minutes |
| Relative Cost | Low | Moderate to high | Moderate |
Table 2: Impact of Accutase treatment duration on surface protein expression
| Treatment Duration | Surface FasL Expression | Surface Fas Receptor Expression | Cell Viability |
|---|---|---|---|
| 10 minutes | Significant decrease | Significant decrease | >90% |
| 30 minutes | Further decrease | Further decrease | >90% |
| 60 minutes | Undetectable | Undetectable | >90% |
| After 20h Recovery | Nearly complete recovery | Nearly complete recovery | >90% |
Recommended Protocols for Insect Cell Detachment
The following protocols are optimized for strongly adherent insect cell lines, particularly Sf9 and Sf21 cells, which require specific handling considerations distinct from mammalian cell systems.
Mechanical Detachment Protocol for Insect Cells
This protocol is adapted for strongly adherent insect cells that require detachment while preserving surface protein integrity for downstream applications.
Materials Required:
- Grace's Insect Medium or other appropriate insect cell culture medium
- Culture vessel with adherent insect cells at appropriate confluency
- Sterile cell scraper or mechanical shaking platform
- Centrifuge tubes
- Hemocytometer or automated cell counter
Procedure:
- Maintain insect cells at 27°C in a non-humidified environment as standard for insect cell culture [1].
- Visually assess cell confluency under microscope. For strongly adherent insect cells, passage at confluency or slightly after when cells naturally begin to pull away from the substrate [1].
- Remove and discard spent culture medium from the culture vessel.
- Gently wash the cell monolayer with pre-warmed calcium- and magnesium-free buffer to remove residual medium.
- For shake-off method: Tighten the cap securely and apply one quick, firm shake using a wrist-snapping motion. Avoid vigorous shaking which may damage cells [1].
- For scraping method: Use a sterile cell scraper to gently but firmly dislodge cells by applying even pressure across the growth surface.
- Immediately add fresh pre-warmed complete growth medium to neutralize the mechanical force effect.
- Transfer the cell suspension to a sterile centrifuge tube and centrifuge at 200 × g for 5-10 minutes.
- Resuspend the cell pellet in fresh medium and count using a hemocytometer or automated cell counter.
- Seed cells at appropriate density for continued culture or experimental use.
Troubleshooting Tips:
- If cells remain adherent after initial attempt, avoid repetitive aggressive scraping which reduces viability.
- For Sf9 cells exhibiting particularly strong adhesion, slight tapping of the flask against the palm may supplement the shaking protocol.
- Mechanical methods typically yield cell clusters rather than single cells; gentle pipetting may help dissociate clusters if single cells are required.
Enzymatic Detachment Protocol for Insect Cells
This protocol utilizes Accutase, recommended as a milder enzymatic alternative to trypsin for insect cell dissociation, though with noted considerations for surface protein integrity.
Materials Required:
- Pre-warmed Accutase or other enzymatic dissociation reagent
- Appropriate insect cell culture medium (e.g., Grace's Insect Medium)
- Balanced salt solution without calcium and magnesium
- Culture vessel with adherent insect cells
- Centrifuge tubes
- Hemocytometer or automated cell counter
Procedure:
- Pre-warm Accutase and complete growth medium to 27°C (standard for insect cell culture) before use [1].
- Remove and discard spent cell culture medium from the culture vessel.
- Wash cells using a balanced salt solution without calcium and magnesium (approximately 2 mL per 10 cm² culture surface area) [1]. Gently add wash solution to the side of the vessel opposite the attached cell layer.
- Remove and discard the wash solution.
- Add pre-warmed Accutase to cover the cell layer (approximately 0.5 mL per 10 cm²) [1].
- Incubate at 27°C for 5-15 minutes. The actual incubation time varies with the cell line and confluency.
- Observe cells under microscope for detachment. If cells are less than 90% detached, increase incubation time, checking every 2-3 minutes.
- When ≥90% of cells have detached, gently tap the vessel to expedite detachment of any remaining cells.
- Add the equivalent of 2 volumes of pre-warmed complete growth medium to neutralize the enzyme.
- Transfer the cells to a centrifuge tube and centrifuge at 200 × g for 5-10 minutes.
- Resuspend the cell pellet in a minimal volume of pre-warmed complete growth medium and remove a sample for counting.
- Seed cells at appropriate density for continued culture.
Critical Considerations:
- Accutase treatment requires 20 hours for full recovery of surface proteins like FasL and Fas receptor; plan experiments accordingly [36].
- Despite surface protein effects, Accutase maintains excellent cell viability (>90%) even with extended incubation up to 90 minutes [36].
- For downstream applications requiring intact surface proteins, consider non-enzymatic alternatives or allow adequate recovery time post-detachment.
Decision Framework for Detachment Method Selection
Diagram 1: Decision framework for detachment method selection (Max Width: 760px)
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Essential research reagents for adherent insect cell detachment
| Reagent/Material | Type | Primary Function | Application Notes |
|---|---|---|---|
| Accutase | Enzymatic | Proteolytic dissociation of adherent cells | Mild enzyme; preserves many surface markers but compromises FasL/Fas receptor [36] |
| TrypLE Express | Enzymatic | Trypsin replacement for cell dissociation | Animal origin-free; direct substitute for trypsin [37] |
| Cell Dissociation Buffer | Non-enzymatic | Chelates calcium and magnesium ions | Preserves surface proteins; ideal for flow cytometry [37] |
| EDTA Solution | Non-enzymatic | Calcium chelation disrupting cell adhesion | Mild; requires mechanical assistance for strongly adherent cells [36] |
| Cell Scraper | Mechanical | Physical dislodgement of adherent cells | Preserves surface proteins; may damage some cells [37] |
| Grace's Insect Medium | Culture medium | Support insect cell growth and maintenance | Formulated for insect cells; more acidic than mammalian media [1] |
The selection between mechanical force and enzymatic reagents for detaching strongly adherent insect cells necessitates careful consideration of experimental goals and downstream applications. Based on current research, the following recommendations are provided:
For maximum surface protein integrity: Utilize mechanical methods (controlled scraping) or non-enzymatic chemical dissociation buffers, particularly when analyzing receptors like FasL and Fas where enzymatic cleavage would compromise results [36].
For high viability and efficient detachment: Employ enzymatic methods with Accutase, which maintains >90% viability even with extended incubation, but allow 20 hours for surface protein recovery before conducting experiments requiring intact membrane proteins [36].
For strongly adherent insect cells: Implement the specific mechanical detachment techniques recommended for insect cells, including the "wrist-snapping motion" for dislodgement, while recognizing that insect cells typically require more vigorous methods than mammalian cells due to their strong adherence under serum-free conditions [1].
Researchers should align their detachment strategy with both immediate experimental needs and long-term culture health, recognizing that method selection significantly influences cellular physiology, protein expression, and ultimately, experimental validity in insect cell-based research systems.
Subculturing, or passaging, is a fundamental technique in cell culture that allows for the continuous maintenance and expansion of cell lines. For researchers working with adherent insect cells, this process is a critical pillar supporting diverse fields, from basic developmental biology to the industrial production of complex proteins, vaccines, and viral vectors using systems like baculovirus expression [5]. Unlike their mammalian counterparts, adherent insect cells present unique challenges; they often attach to substrates with exceptional tenacity, especially under serum-free conditions commonly used in bioproduction [1]. Furthermore, their optimal growth occurs at a non-mammalian 27°C, typically without CO₂ exchange or a humidified environment [1]. This application note provides a detailed, step-by-step protocol for the subculturing of adherent insect cells, framed within the context of ensuring experimental reproducibility and viability in both academic and industrial drug development workflows.
Materials and Equipment
Research Reagent Solutions and Essential Materials
The following table details the key reagents and materials required for the successful subculturing of adherent insect cells.
Table 1: Essential Materials for Subculturing Adherent Insect Cells
| Item | Function and Specification |
|---|---|
| Growth Medium | Culture medium specifically formulated for insect cells, such as Grace's medium. These are typically more acidic than mammalian media [1]. |
| Dissociation Reagent | A trypsin-based solution (e.g., 0.05 mg/ml VMF trypsin) or a recombinant enzyme alternative like TrypLE, used to detach cells from the culture surface [5]. |
| Balanced Salt Solution | A divalent cation-free solution, such as PBS without calcium and magnesium, used to wash cells and remove traces of serum that would inhibit trypsin [1] [5]. |
| Complete Growth Medium | Growth medium containing serum or other supplements that inactivate the dissociation reagent and provide nutrients for renewed growth [1]. |
| Cryoprotectant (DMSO) | Used for cryopreservation of cell stocks, typically at a concentration of 5-10% to prevent ice crystal formation [4]. |
Necessary Equipment
- Laminar flow hood or biological safety cabinet [5]
- Inverted phase contrast microscope [5]
- Refrigerated incubator set to 27°C (non-humidified) [1]
- Centrifuge [1]
- Mechanical pipetting device, serological pipettes, and pipette tips [5]
- Hemocytometer or automated cell counter (e.g., Invitrogen Countess) [1]
- 25-cm² tissue culture flasks and centrifuge tubes [5]
The following diagram outlines the complete subculturing workflow for adherent insect cells, highlighting the critical decision points and steps.
Diagram 1: Subculturing workflow for adherent insect cells.
Step-by-Step Protocol
Pre-subculture Assessment and Preparation
Principle: Passage cells during the log phase of growth, before they reach confluency, unless the specific cell line is strongly adherent. For strongly adherent cells, passaging at confluency or slightly after may be easier as cells begin to pull away from the flask [1].
- Observation: Examine cells under an inverted phase contrast microscope. Healthy cells should appear refractive. The medium should be clear; cloudiness may indicate bacterial contamination [5].
- Timing: Passage before confluency for most lines. Strongly adherent cells can be passaged at confluency [1].
- Preparation: Wipe down the laminar flow hood with 70% ethanol. Pre-warm the dissociation reagent, balanced salt solution, and complete growth medium to room temperature. Label new culture flasks with the cell line, date, and passage number [1] [5].
Washing and Trypsinization
Principle: The wash step removes residual serum, calcium, and magnesium that inhibit trypsin. The dissociation reagent then breaks the cell-substrate and cell-cell attachments.
- Remove Media: Aspirate and discard the spent cell culture media from the culture vessel [1].
- Wash Cells: Gently add a balanced salt solution without calcium and magnesium (approximately 2 mL per 10 cm² surface area) to the side of the vessel opposite the cell layer. Rock the vessel gently, then remove and discard the wash solution [1].
- Add Dissociation Reagent: Add pre-warmed dissociation reagent (e.g., trypsin or TrypLE) to just cover the cell layer (approx. 0.5 mL per 10 cm²). Gently rock the vessel for complete coverage [1].
- Incubate and Monitor: Incubate the vessel at room temperature for approximately 2 minutes. Actual incubation time varies; observe under a microscope every 30 seconds for cell detachment [1].
Cell Detachment and Dissociation
Principle: Neutralizing the trypsin and mechanically dispersing the cells ensures a single-cell suspension for accurate counting and uniform reseeding.
- Confirm Detachment: When ≥90% of cells appear rounded up and detached, tilt the vessel to allow cells to drain [1].
- Mechanical Force for Stubborn Cells: For insect cells that attach very tightly, a sharp tap with the palm of the hand may be necessary. Avoid vigorous shaking, which can damage cells [1] [5].
- Neutralize Trypsin: Add the equivalent of 2 volumes of pre-warmed complete growth medium (twice the volume of dissociation reagent used) to inactivate the enzyme. Disperse the medium by pipetting over the cell layer surface several times to ensure a single-cell suspension [1].
Cell Harvesting and Counting
Principle: Concentrating cells via centrifugation allows for the removal of the spent dissociation reagent and precise preparation of a new cell suspension at the optimal density.
- Transfer and Centrifuge: Transfer the cell suspension to a conical tube and centrifuge at 200 x g for 5–10 minutes [1].
- Resuspend: Carefully decant the supernatant and resuspend the cell pellet in a minimal volume of fresh, pre-warmed complete growth medium [1].
- Count and Assess Viability: Remove a sample for counting. Determine the total cell count and percent viability using a hemocytometer and Trypan Blue exclusion method or an automated cell counter. Viability should be greater than 90% at the time of subculturing [1] [4].
Reseeding and Incubation
Principle: Seeding cells at the correct density is crucial for promoting rapid attachment and subsequent logarithmic growth.
- Dilute Cell Suspension: Using the cell count, dilute the cell suspension with complete growth medium to achieve the recommended seeding density for your specific insect cell line.
- Seed New Vessels: Pipet the appropriate volume of the diluted cell suspension into new, labeled culture vessels.
- Incubate: Place the new cultures in a 27°C incubator. Insect cells do not require a humidified environment or CO₂ exchange. If using non-vented flasks, loosen the caps to allow for gas exchange [1].
Critical Parameters and Data Presentation
Successful subculturing relies on optimizing key parameters. The following tables consolidate quantitative data and observations to guide researchers.
Table 2: Key Incubation and Physical Parameters for Adherent Insect Cells
| Parameter | Optimal Condition | Rationale & Considerations |
|---|---|---|
| Incubation Temp. | 27°C | Standard for most insect cell lines; a controlled environment is recommended over room temperature [1]. |
| CO₂ / Humidity | Not required | Unlike mammalian cells, insect cell culture systems do not rely on sodium bicarbonate buffering [1]. |
| Detachment Force | Sharp tap (if needed) | Essential for strongly adherent cells grown in serum-free media. Avoid vigorous shaking [1]. |
| Confluency at Passage | Pre-confluency (most); At confluency (strongly adherent) | Pre-confluency passage requires more force. Repeated passaging at incorrect densities decreases viability and doubling times [1]. |
| Centrifugation Speed | 200 x g for 5-10 min | Standard speed for pelleting cells; can be varied based on cell type [1]. |
Table 3: Troubleshooting Common Issues in Insect Cell Subculture
| Observed Problem | Potential Cause(s) | Recommended Solution(s) |
|---|---|---|
| Slow or No Detachment | Insufficient trypsin incubation; overly adherent cells; inactive trypsin. | Increase incubation time; apply a sharp, quick tap to the flask; ensure fresh, properly stored reagents [1] [5]. |
| Poor Cell Viability Post-Seeding | Over-typsinization; incorrect centrifugation force/speed; old or contaminated medium. | Monitor detachment closely and neutralize promptly; verify correct centrifugation protocol; use fresh, sterile medium [4]. |
| Decreased Doubling Times | Repeated passaging at incorrect densities (too low or too high). | Adhere to recommended confluency levels for passaging specific to the cell line [1]. |
| Culture Contamination | Break in aseptic technique; contaminated reagent. | Discard culture; review sterile technique; quality-check all reagents [4] [5]. |
The subculturing of adherent insect cells is a precise technique that, when mastered, ensures the health and reliability of a critical research tool. This detailed walkthrough underscores that success hinges not only on following the mechanical steps of washing, trypsinization, and reseeding but also on a deep understanding of the unique biology of insect cells. Key differentiators from mammalian cell culture—such as temperature, the frequent need for mechanical dislodgement, and strict adherence to passaging at the correct density—are paramount. By integrating these specific considerations and maintaining rigorous aseptic technique, researchers and drug development professionals can achieve consistent, high-yield results, thereby underpinning the integrity of their scientific and commercial applications in the field of biotechnology.
Adapting to Serum-Free and Animal Component-Free Media for Clinical Applications
The transition from traditional serum-supplemented media to serum-free (SFM) and animal component-free (ACF) media represents a critical advancement in cell culture technology for clinical applications. This shift is driven by the need for safer, more standardized, and reproducible manufacturing processes for cell therapies, vaccines, and other biological products [39] [40]. While fetal bovine serum (FBS) has been a culture mainstay for decades, its use in clinical manufacturing is hampered by significant scientific and ethical concerns [41].
FBS is a biologically undefined substance with a complex composition that varies from batch to batch, leading to inconsistencies in experimental and production outcomes [42] [41]. Furthermore, it carries the risk of introducing adventitious agents, such as viruses and prions, and contains immunogenic xeno-proteins that can elicit unwanted immune responses in patients [43] [41]. From an ethical standpoint, the collection of FBS raises substantial animal welfare concerns [41]. Serum-free and animal component-free media address these issues by offering a defined, consistent, and safer platform for cultivating cells intended for human therapies [39] [40].
This application note provides detailed guidance on adapting adherent cell cultures to these advanced media, framed within the context of clinical manufacturing. It includes performance comparison data, step-by-step protocols, and a special focus on considerations for adherent insect cells, a critical production system for viral vectors and recombinant proteins.
Understanding Media Types and Their Clinical Advantages
Navigating the terminology of modern cell culture media is essential for selecting the right product for clinical applications. The table below defines key terms and summarizes their importance.
Table 1: Classification and Clinical Relevance of Advanced Culture Media
| Media Type | Definition | Clinical & Manufacturing Advantages |
|---|---|---|
| Serum-Free (SFM) | Contains no unpurified serum, but may contain purified animal-derived components (e.g., growth factors, lipids) [42]. | Reduces batch-to-batch variability; lowers risk of contamination from serum-borne pathogens [40]. |
| Animal Component-Free (ACF) | Contains no components of animal origin in the final formulation or manufacturing process [43]. | Eliminates xeno-immunogenic risks; simplifies regulatory approval for cell and gene therapies [39]. |
| Xeno-Free (XF) | A subset of ACF; implies no non-human animal components are used [43]. | Essential for clinical applications to prevent immune reactions to non-human antigens [41]. |
| Chemically Defined (CDM) | All components are known, pure chemicals of defined concentration; no biological or animal-derived materials [43]. | Maximizes reproducibility and process control; ideal for GMP manufacturing and regulatory compliance [40] [44]. |
The adoption of these defined media is supported by a strong regulatory push. For instance, the first US FDA approval for a serum-free culture medium for mesenchymal stem cells (MSCs) was recently achieved, highlighting the growing global shift toward safer, standardized cell therapy solutions [39].
However, researchers must be aware that commercial product labels can sometimes be misleading. A 2025 study found that some commercially available "SFM" contained significant levels of human platelet lysate (hPL) components, effectively reclassifying them as hPL-supplemented media rather than true SFM [42]. This underscores the importance of thorough supplier qualification and, where necessary, in-house verification of critical media components.
Performance Comparison and Selection Criteria
Selecting the appropriate medium requires a careful balance of performance, cost, and regulatory compliance. The following table synthesizes quantitative and qualitative data from comparative studies.
Table 2: Performance and Economic Comparison of Culture Media Supplements
| Parameter | Fetal Bovine Serum (FBS) | Human Platelet Lysate (hPL) | Serum-Free Media (SFM) |
|---|---|---|---|
| Composition | Undefined, complex mixture of >1,800 proteins [41]. | Defined but variable; rich in human growth factors [42]. | Defined, though some may contain purified proteins [42]. |
| Batch Variability | High; a major source of experimental irreproducibility [41]. | Moderate to high; depends on donor pool and manufacturing [42]. | Low (theoretically), but requires rigorous vendor qualification [42]. |
| Cell Growth (MSC Expansion) | Standard, but may induce non-physiological phenotypes [41]. | Consistently supports high cell growth [42]. | Variable performance; some support growth well, others poorly [42]. |
| Cost | Fluctuating supply and cost [41]. | More cost-effective than SFM [42]. | Significantly higher cost than hPL and FBS [42]. |
| Regulatory & Safety Profile | High risk; xenogenic, potential for contaminants [41]. | Lower risk; human-derived, but requires pathogen testing [43]. | Ideal; low contamination risk, non-immunogenic [39] [41]. |
This comparative analysis indicates that while SFM and CDM offer the best regulatory and safety profiles for clinical work, their performance must be validated on a cell-line-specific basis. Human Platelet Lysate (hPL) presents a viable, more cost-effective xeno-free alternative that generally supports robust cell growth, making it a practical option for many clinical-scale expansions [42].
General Protocol for Adapting Adherent Mammalian Cells
Adapting cells to a new medium requires a methodical approach to avoid shocking the cells and to ensure long-term stability. The following protocol is recommended for transitioning adherent mammalian cells from serum-containing to serum-free or animal component-free media.
Materials and Reagents
- Base Media: The new, validated serum-free or chemically defined medium.
- Serum-Containing Medium: The original growth medium.
- Cell Line: A healthy, log-phase culture of the adherent cells to be adapted.
- Culture Vessels: Appropriate flasks or plates, sometimes requiring pre-coating with extracellular matrix proteins (e.g., laminin, fibronectin) for SFM [42] [44].
- Detachment Reagent: An animal-origin-free enzyme solution, such as recombinant trypsin (TrypLE) [1].
- Phosphate-Buffered Saline (PBS): Without calcium and magnesium.
- Additional Supplements: Specific growth factors or lipids as required by the new medium formulation.
Sequential Adaptation Workflow
The adaptation process should be gradual, systematically increasing the proportion of the new medium over several passages. The following diagram illustrates this workflow.
Detailed Step-by-Step Procedure
- Initial Seeding: Begin the adaptation process when your cells are in the log phase of growth and have viability greater than 90% [1]. Subculture the cells as you normally would, using your standard detachment reagent. After centrifugation, resuspend the cell pellet in a mixture of 75% original serum-containing medium and 25% new SFM/ACF medium. Seed the cells at a density that is standard for the cell line [4] [45].
- Monitoring and Subsequent Passaging: Monitor the cells daily for changes in morphology, confluency, and medium color (if phenol red is used). Passage the cells while they are still in the log phase, before they reach 100% confluency [1] [45]. At the next passage, resuspend the cells in a 50:50 mixture of old and new media.
- Progression to Full Adaptation: Continue passaging the cells, each time increasing the proportion of the new SFM/ACF medium. A typical sequence is 75% new medium, followed by 100% new medium.
- Assessment of Adaptation Success: After at least one passage in 100% new medium, critically assess the adaptation. Key metrics include:
- Growth Kinetics: Compare the population doubling time to that in the original medium. A slight change is common, but a significant increase indicates a problem.
- Viability: Should remain consistently above 90% [1].
- Morphology: Cells should maintain their characteristic shape and appearance under a light microscope.
- Function: For the final clinical application, ensure the cells retain their critical functionality (e.g., differentiation potential, protein production, etc.) [40].
Special Considerations for Adherent Insect Cells
Insect cell culture systems, crucial for baculovirus-based production of viral vectors and recombinant proteins, have distinct requirements that must be considered during adaptation to SFM.
Key Differences from Mammalian Systems
- Culture Environment: Insect cells are typically maintained at 27°C in a non-humidified, non-CO₂ environment [1].
- Medium Acidity: Growth media for insect cells, such as Grace's medium, are often more acidic than those for mammalian cells [1].
- Cell Adhesion: Under serum-free conditions, insect cells can attach very tightly to the substrate, making them more difficult to detach during passaging [1].
Modified Subculture Protocol for Insect Cells
The general adaptation workflow in Section 4.2 applies, but the subculture technique itself must be modified.
- When to Subculture: Passage insect cells at confluency or slightly after, when they may start to pull away from the flask bottom. Passaging before confluency can require excessive force to dislodge them and lead to decreased viability and doubling times [1].
- Detachment: After rinsing with a balanced salt solution and incubating with a dissociation reagent, insect cells may need a physical impetus to detach. Tighten the cap and give the flask one quick, firm shake using a "wrist-snapping motion." Do not shake vigorously, as this can damage the cells [1].
- Post-Detachment Processing: Once detached, proceed with centrifugation and resuspension in the new medium mixture as described for mammalian cells.
Analytical Methods for Verifying Adaptation Success
Once cells are established in the new medium, rigorous testing is required to confirm they meet the necessary standards for clinical use. The following diagram and table outline a verification workflow and key methods.
Table 3: Key Analytical Methods for Post-Adaptation Verification
| Analysis Category | Specific Method | Measured Outcome | Clinical Rationale |
|---|---|---|---|
| Proliferation & Health | Automated cell counting with Trypan blue exclusion [1] [4]. | Growth curve, population doubling time, viability %. | Ensures cells expand robustly and maintain health in the new medium. |
| Cell Composition | ELISA for human proteins (e.g., fibrinogen, glycocalicin) [42]. | Verification of media composition and absence of residual contaminants. | Confirms the "serum-free" or "xeno-free" status of the culture. |
| Phenotype & Identity | Flow cytometry for cell-specific surface markers (e.g., CD44 for MSCs) [42]. | Confirmation of cellular identity and lack of phenotypic drift. | Critical for cell therapies to ensure the product contains the correct, functional cell type. |
| Functionality | Cell-specific potency assay (e.g., differentiation, cytokine secretion, virus production). | Demonstration of desired biological activity. | Links the manufacturing process to the intended biological function and clinical effect. |
| Safety | Sterility, mycoplasma, and endotoxin testing per pharmacopoeial methods. | Confirmation of a contamination-free product. | Mandatory for release of any cell-based product for clinical use. |
The Scientist's Toolkit: Essential Reagents for Serum-Free Transition
Successfully establishing a culture in serum-free or animal component-free media often requires specific reagents that replace the functions of serum. The following table details essential components for your toolkit.
Table 4: Essential Reagents for Serum-Free and Animal Component-Free Cell Culture
| Reagent / Solution | Function | Example & Notes |
|---|---|---|
| Basal Defined Medium | Provides salts, vitamins, energy sources, and buffers. | DMEM/F12, RPMI 1640, or proprietary SFM formulations. Must be validated for your cell type. |
| Attachment Factors | Promotes cell adhesion and spreading in place of serum proteins. | Human-derived or recombinant proteins like fibronectin, laminin, or Collagen Type IV [44]. |
| Carrier Proteins | Binds lipids, hormones, and vitamins; stabilizes other proteins. | Human Serum Albumin (HSA) or recombinant albumin is a common, critical component [44]. |
| Growth Factors | Stimulates cell proliferation and maintains specific phenotypes. | Recombinant human proteins like EGF, bFGF, IGF-1, and PDGF are essential [44] [41]. |
| Lipids & Lipid Carriers | Provides cholesterol, fatty acids, and precursors for membranes. | A chemically defined lipid concentrate or complexed with cyclodextrin [44]. |
| Enzymatic Detachment Reagent | Dissociates adherent cells for passaging without animal components. | Recombinant trypsin substitutes like TrypLE, which is animal-origin-free [1]. |
| Cryopreservation Medium | Protects cells during freeze-thaw in the absence of serum. | Pre-formulated ACF solutions or defined media with cryoprotectants like DMSO [4]. |
The migration to serum-free and animal component-free media is no longer a niche pursuit but a fundamental requirement for robust, safe, and compliant clinical manufacturing. While the adaptation process demands careful planning, execution, and validation, the benefits of a defined, consistent, and safe culture environment are undeniable. By following structured protocols, leveraging appropriate analytical tools, and understanding the specific needs of different cell systems like adherent insect cells, researchers and developers can successfully navigate this transition. This, in turn, accelerates the development of reliable and effective advanced therapeutic medicines.
Solving Common Challenges: A Troubleshooting Guide for Robust Cultures
Maintaining robust insect cell cultures is fundamental to successful recombinant protein production and baculovirus-based research. A prevalent challenge leading to decreased cell viability and extended doubling times is the suboptimal timing of passaging, specifically harvesting cells at incorrect confluency intervals. This application note delineates the critical parameters for the maintenance of adherent insect cell lines, such as Sf9, Sf21, and High Five, with a focused protocol on determining and adhering to the correct passaging confluency. By integrating quantitative culture parameters and a detailed methodological workflow, we provide a standardized approach to circumvent common pitfalls, thereby ensuring consistent cellular health and experimental reproducibility.
Insect cell lines, particularly those derived from Spodoptera frugiperda (Sf9 and Sf21), are cornerstones of the baculovirus expression vector system (BEVS) for multiprotein expression [17] [10]. The health of these cultures is paramount, as it directly impacts recombinant protein yield, viral titer, and overall experimental success. A frequently encountered issue in laboratory practice is the gradual decline in culture health, manifesting as decreased percent viability and increased population doubling times. Empirical evidence from experienced laboratories indicates that a primary contributor to this decline is the repeated passaging of cells at inappropriate confluency levels [1] [17].
Passaging adherent insect cells either too early (before confluency) or too late (significantly post-confluency) imposes stress, leading to phenotypic and genotypic instability. Cells passaged pre-confluency require excessive mechanical force for detachment, causing physical damage, while cells passaged post-confluency suffer from nutrient depletion and contact inhibition [1]. This application note, framed within a broader thesis on subculturing adherent insect cells, establishes a definitive protocol to identify and maintain the optimal passaging window, thereby preserving the integrity of cellular research for scientists and drug development professionals.
Results and Data Presentation
Quantitative Culture Parameters for Common Insect Cell Lines
The table below summarizes the critical quantitative parameters for maintaining robust growth in adherent cultures of commonly used insect cell lines, based on a 3-to-4-day subculture schedule [46].
Table 1: Adherent Culture Parameters for Insect Cell Lines
| Cell Line | Incubation Temperature (°C) | Recommended Seeding Density (viable cells/cm²) | Doubling Time (Hours) | Recommended Passaging Confluency |
|---|---|---|---|---|
| Sf9 | 26 - 28°C | 6 - 7 x 10E4 | 24 - 30 | 70-90% |
| Sf21 | 26 - 28°C | 6 - 7 x 10E4 | 24 - 30 | 70-90% |
| High Five | 26 - 28°C | 2 - 5 x 10E4 | ~24 | 70-90% |
| D.mel-2 | 26 - 28°C | 5 - 6 x 10E4 | 18 - 24 | 70-90% |
Consequences of Incorrect Passaging Confluency
Deviating from the optimal passaging confluency of 70-90% has direct and detrimental effects on cell health, as synthesized from empirical observations [1] [17].
Table 2: Impact of Suboptimal Passaging on Cell Health
| Passaging Condition | Observed Morphological & Behavioral Signs | Consequences for Cell Health |
|---|---|---|
| Too Early (<70% confluency) | Cells are strongly adherent, requiring sharp mechanical force to dislodge. Monolayer appears sparse. | Decreased doubling times, decreased viability, reduced ability to attach in subsequent passages. Considered unhealthy. |
| Too Late (>90%-100% confluency) | Cells begin to detach from the monolayer spontaneously. Medium may become acidic (yellow if phenol red is used). | Decreased doubling times, decreased viability, and reduced attachment ability due to nutrient depletion and waste accumulation. |
| Repeatedly at Densities Past Confluency | Overgrowth, increased cellular debris, and drop in medium pH. | A significant and often irreversible decline in all growth parameters, leading to an unusable culture. |
Experimental Protocols
Detailed Protocol: Passaging Adherent Insect Cells at Correct Confluency
This protocol is optimized for adherent Sf9 and Sf21 cells but is applicable to other adherent insect cell lines with adjustments to seeding density as specified in Table 1.
Materials and Reagents
- Insect Cells: Adherent culture of Sf9, Sf21, or other insect cell line at 70-90% confluency.
- Growth Medium: Pre-warmed serum-free medium (e.g., Sf-900 II SFM for Sf cells) or serum-supplemented medium (e.g., Grace's insect medium with 10% heat-inactivated FBS) [46].
- Dissociation Reagent: Pre-warmed, non-enzymatic (e.g., TrypLE) or enzymatic dissociation reagent.
- Balanced Salt Solution: Sterile, without calcium and magnesium (e.g., Dulbecco's Phosphate Buffered Saline - DPBS).
- Culture Vessels: T-flasks or other appropriate treated plasticware.
- Centrifuge Tubes, Pipettes, and other standard cell culture equipment.
Step-by-Step Procedure
- Monitoring and Timing: Routinely monitor cell confluency using an inverted microscope. Initiate passaging only when the culture reaches 70-90% confluency [1].
- Preparation: Pre-warm the complete growth medium and dissociation reagent to room temperature (or 27°C).
- Medium Removal: Aseptically remove and discard the spent culture medium from the adherent cell layer.
- Wash: Gently rinse the cell layer with a balanced salt solution (approximately 2 mL per 10 cm²) to remove residual serum and calcium, which can inhibit dissociation. Remove and discard the wash solution.
- Dissociation: Add enough pre-warmed dissociation reagent to cover the cell layer (approximately 0.5 mL per 10 cm²). Gently rock the vessel to ensure complete coverage.
- Incubation: Incubate the culture vessel at room temperature for approximately 2-5 minutes. The actual time varies; observe cells under a microscope for detachment.
- Cell Detachment: When ≥90% of cells have detached and rounded up, tap the vessel firmly if necessary. Avoid vigorous shaking, as this can damage cells [1].
- Neutralization: Add the equivalent of 2 volumes of pre-warmed complete growth medium to neutralize the dissociation reagent. Disperse the medium by pipetting over the cell layer surface several times to obtain a single-cell suspension.
- Cell Counting and Seeding: Transfer the cell suspension to a sterile tube. Determine the cell count and viability using a hemocytometer or automated cell counter with Trypan blue exclusion. Dilute the cell suspension with fresh, pre-warmed medium to the recommended seeding density (Refer to Table 1) and pipet the appropriate volume into a new culture vessel.
- Incubation: Cap the flask loosely to allow for gas exchange and incubate cells at 27°C in a non-humidified, non-CO₂ environment [46] [1].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Adherent Insect Cell Culture
| Reagent / Material | Function & Rationale | Example Products & Notes |
|---|---|---|
| Serum-Free Insect Medium | Provides nutrients and buffering system (pH ~6.2). Avoids batch variability of serum and supports high-density growth. | Sf-900 II SFM, Sf-900 III SFM, Express Five SFM. Some contain surfactants like Pluronic F-68 to reduce shear stress [46] [47]. |
| Non-Enzymatic Dissociation Reagent | Gently disrupts cell attachments to the substrate without damaging surface proteins, which can occur with trypsin. | TrypLE Select. Preferred for insect cells which attach very tightly under serum-free conditions [1]. |
| Pluronic F-68 | A non-ionic surfactant that protects cell membranes from shear stress caused by fluid dynamics and pipetting. | Add to a final concentration of 0.1% in media that do not already contain it, especially for suspension adaptation [46] [47]. |
| Heat-Inactivated Fetal Bovine Serum (FBS) | Provides a broad spectrum of growth factors, hormones, and attachment factors. | Used at 10% in media like Grace's supplemented or Schneider's Drosophila medium. Heat-inactivation removes complement proteins [46]. |
Discussion
The precise timing of cell passaging is not merely a procedural step but a critical determinant of long-term culture stability. The data and protocol presented herein underscore that adherence to a 70-90% confluency threshold is a simple yet effective strategy to mitigate the common issue of declining viability and increasing doubling times. This practice minimizes mechanical and metabolic stress, allowing cells to remain in a consistent, logarithmic growth phase [1].
For researchers troubleshooting unhealthy cultures, the first corrective action should be a rigorous audit of passaging records and confluency at subculture. It is noteworthy that Sf9 cells often demonstrate higher tolerance to variations in culture conditions and high cell densities compared to Sf21 cells, making them a more robust choice for routine virus amplification and protein expression [17] [10]. Furthermore, the use of specialized reagents, such as non-enzymatic dissociation agents and media-containing surfactants detailed in Table 3, provides an additional layer of protection for maintaining cell integrity throughout the passaging process.
In conclusion, by integrating the quantitative guidelines, visual monitoring skills, and standardized protocols outlined in this application note, researchers can establish a reproducible and reliable insect cell culture system, forming a solid foundation for advanced applications in protein expression and drug development.
Managing Contamination in a Non-Humidified, Room-Temperature Incubation Setup
The use of non-humidified, room-temperature incubation for adherent insect cell culture presents distinct advantages in simplicity and cost-effectiveness for research and bioprocessing applications. However, this environment introduces unique contamination control challenges that differ significantly from standard mammalian cell culture systems. Insect cells, such as Sf9, Sf21, and High Five, are maintained at 26-28°C without CO₂ exchange or humidity control [46]. While eliminating the humidification requirement reduces fungal and bacterial growth potential, the non-sterile ambient air exposure and absence of standardized incubator environments create vulnerabilities to airborne contaminants and environmental fluctuations. This application note outlines evidence-based protocols for managing contamination within this specific context, supporting the broader research thesis on optimizing adherent insect cell subculturing methodologies.
Material and Reagent Solutions
Table 1: Essential Reagents for Contamination Management in Insect Cell Culture
| Reagent/Category | Specific Examples | Function in Contamination Control |
|---|---|---|
| Cell Culture Media | Sf-900 II SFM, Sf-900 III SFM, Express Five SFM [46] | Serum-free formulations reduce nutrient source for contaminants |
| Detachment Reagents | Trypsin-based solutions, Non-enzymatic dissociation buffers [1] [48] | Maintain cell integrity during subculturing to prevent vulnerability |
| Surface Disinfectants | 70% ethanol, 10% bleach solutions [49] [50] | Decontaminate work surfaces and equipment |
| Antibiotics/Antimycotics | Penicillin-Streptomycin (Pen/Strep) [46] | Short-term suppression of bacterial/fungal growth; use sparingly |
| Cell Culture Additives | Pluronic F-68 (for suspension cultures) [46] | Protect cells from shear stress without promoting contamination |
Understanding the Unique Incubation Environment and Associated Risks
Insect cell cultures for recombinant protein production and baculovirus expression systems are typically maintained at 27°C ± 1°C in ambient, non-humidified air [46] [1]. Unlike mammalian cultures requiring 37°C and 5% CO₂, this simplified environment lacks the built-in contamination barriers of humidified incubators with gas control. The primary contamination pathways in this setup include:
- Airborne contaminants due to direct exposure to room air without HEPA filtration
- Personnel-borne microorganisms introduced during handling
- Compromised reagents and media given the open bench-top nature of the system
- Cross-contamination between cell lines when multiple cultures are handled simultaneously [49] [50]
Insect cells present additional vulnerabilities because they are "much more fragile than a lot of mammalian cell lines" and suffer damage from both overgrowth and over-splitting [46]. This fragility increases susceptibility to contamination, as stressed cells have reduced defense mechanisms. Furthermore, insect cell culture media with its characteristic acidic pH (6.0–6.4) [46] may initially inhibit some bacterial contaminants but favor acid-tolerant microorganisms.
Comprehensive Prevention Protocols
Aseptic Technique Specialization for Non-Humidified Systems
- Pre-handling preparation: Wipe down all work surfaces with 70% ethanol before initiating procedures [5] [49]. Inspect cultures for early signs of contamination using an inverted phase contrast microscope [5].
- Single-cell line policy: Work with only one insect cell line at a time to prevent cross-contamination [50]. Thoroughly clean the biosafety cabinet with 70% alcohol before and after introducing a new cell line [50].
- Personal protective equipment: Wear proper gloves and lab coats, and wash hands when entering or leaving the lab [49]. Avoid talking during critical manipulations to minimize aerosol formation [45].
Environmental Control and Monitoring
- Temperature regulation: Maintain incubation temperature at 27°C ± 1°C [46] [1]. Avoid room temperature fluctuations by using dedicated, temperature-stable incubators rather than open bench tops. Prolonged exposure to temperatures above 29°C causes insect cell death [46].
- Air quality management: Implement regular cleaning of incubator interiors with laboratory disinfectants even in non-humidified systems [51]. Consider portable HEPA filtration units for rooms used for insect cell culture.
- Vessel integrity: Ensure proper sealing of culture vessels while allowing for minimal gas exchange. Tighten caps before mechanical manipulation of flasks to avoid contamination [1].
Reagent and Media Management
- Media formulation: Use insect-specific media such as Sf-900 II SFM or Express Five SFM [46]. These are specifically formulated with the appropriate pH and osmolarity (340 μOsM) for insect cells [46].
- Antibiotic usage: Limit antibiotic and antimycotic use to short-term applications only [51]. Their continuous use encourages resistant strains and can hide low-level contamination, particularly mycoplasma [48] [51].
- Sterile technique: Use sterile pipets only once for transfers from medium bottles to avoid contamination problems [5]. Aliquot reagents to minimize repeated exposure to potential contaminants [45].
Procedural Safeguards During Subculturing
Adherent insect cells require special handling during subculturing due to their tight attachment, particularly under serum-free conditions [1]. The mechanical force needed for detachment must be balanced against contamination risk:
- Optimal passage timing: Passage insect cells at confluency or slightly after when they begin to pull away from the flask surface [1]. Subculturing before confluency requires more mechanical force and can decrease viability, increasing contamination susceptibility.
- Mechanical detachment: For strongly adherent cells, strike the flask sharply on the side with the palm two to three times to loosen cells after refrigeration [5]. Avoid vigorous shaking that damages cells and creates microaerosols.
- Post-subculture handling: After passaging, return cultures immediately to the controlled 27°C environment. Document passage information including date, cell line designation, and medium lot in a record book [5].
Contamination Detection and Identification
Regular monitoring is essential for early contamination detection. The following workflow provides a systematic approach for identifying and responding to potential contamination events in insect cell cultures:
Table 2: Contamination Identification Guide for Insect Cell Cultures
| Contaminant Type | Visual Signs | Microscopic Indicators | pH Changes |
|---|---|---|---|
| Bacteria | Cloudy/turbid medium, surface film [51] | Tiny, moving granules between cells [51] | Rapid drop (yellow) [51] [50] |
| Yeast | Turbid medium, possible odor [50] | Ovoid/spherical particles that may bud [51] | Stable initially, then increases [51] |
| Mold | Floating mat-like structures, possible odor [50] | Thin, filamentous mycelia [51] | Stable initially, then increases [51] |
| Mycoplasma | No visible change [51] | No obvious change; requires specialized staining [51] [50] | Minimal change |
Decontamination Protocols for Compromised Cultures
When irreplaceable adherent insect cell cultures become contaminated, decontamination may be attempted following this structured protocol:
Antibiotic/Antimycotic Sensitivity Testing
For confirmed bacterial or fungal contamination in critical cultures:
- Cell preparation: Dissociate, count, and dilute contaminated cells in antibiotic-free medium to concentration used for regular passage [51].
- Dose optimization: Dispense cell suspension into multi-well plates and add antibiotics/antimycotics in a range of concentrations [51].
- Toxicity assessment: Observe cells daily for signs of toxicity (sloughing, vacuoles, decreased confluency, rounding) for 2-3 days [51].
- Treatment application: Culture cells for 2-3 passages using the antibiotic at a concentration one- to two-fold lower than the toxic concentration [51].
- Cure verification: Culture cells in antibiotic-free medium for 4-6 passages to confirm elimination of contamination [51].
System-Wide Decontamination
Following any contamination event:
- Isolate contaminated cultures immediately from other cell lines [51].
- Decontaminate incubators and laminar flow hoods with appropriate laboratory disinfectants [51].
- Check HEPA filters in biosafety cabinets if available [51].
- Autoclave all contaminated materials when possible to prevent spread [49].
Effective contamination management in non-humidified, room-temperature incubation systems for adherent insect cells requires specialized approaches distinct from mammalian cell culture. By implementing these targeted protocols—emphasizing strict aseptic technique, environmental monitoring, and early detection—researchers can maintain robust, contamination-free insect cell cultures. These practices support the reliability of downstream applications in recombinant protein production and virology, forming a critical component of successful adherent insect cell research methodologies.
Optimizing Split Ratios and Seeding Density for Sf9 vs. Sf21 Cells
Within the broader scope of research on subculturing adherent insect cells, the selection of appropriate split ratios and seeding densities is a fundamental determinant of experimental success. The Spodoptera frugiperda cell lines, Sf9 and Sf21, are cornerstones of recombinant protein production, vaccine development, and basic research using the baculovirus expression vector system (BEVS) [17] [14]. While these cell lines share a common origin, their distinct growth kinetics and physical characteristics necessitate optimized, separate subculturing protocols. This application note provides a detailed, comparative guide to the routine maintenance of Sf9 and Sf21 cells, framing these practical procedures within the critical context of ensuring high cell viability and recombinant protein yield.
Comparative Cell Line Characteristics
Understanding the inherent differences between Sf9 and Sf21 cells is a prerequisite for optimizing their culture conditions. The table below summarizes their key characteristics as derived from both commercial and research sources [17] [52].
Table 1: Comparative Characteristics of Sf9 and Sf21 Cell Lines
| Characteristic | Sf9 Cells | Sf21 Cells |
|---|---|---|
| Origin | Subclone of IPLB-Sf21-AE [17] | Parental ovarian cell line from Spodoptera frugiperda [52] |
| Morphology | Small, regular, uniform size; forms exceptional monolayers [52] | Spherical, unequal sizes, granular appearance [52] |
| Growth Rate | Faster [17] | Slower [17] |
| Tolerance | More tolerant to high densities and shear stress in suspension [17] | Less tolerant to condition variation [17] |
| Typical Doubling Time | ~18-20 hours [53] | ~18-20 hours [53] |
| Preferred Application | Virus amplification and high-yield protein production [17] | Plaque assays and initial virus titration [17] |
Optimized Subculture Parameters
Based on established culturing practices, the following parameters are recommended for the routine passage of Sf9 and Sf21 cells. Adherence to these guidelines promotes consistent, healthy growth and prevents the decline of culture health.
Quantitative Seeding and Splitting Guide
The following table provides the quantitative framework for subculturing both adherent and suspension cultures of Sf9 and Sf21 cells.
Table 2: Optimized Split Ratios and Seeding Densities for Sf9 and Sf21 Cells
| Parameter | Optimal Range for Sf9 & Sf21 | Additional Context & Recommendations |
|---|---|---|
| Seeding Density for Maintenance | ( 3.0 \times 10^5 ) cells/mL [52] | This density is suitable for starting suspension cultures in shaker flasks. |
| Adherent Culture Seeding Density | ( 5.0 \times 10^4 ) viable cells/cm² [52] | Used for creating monolayers in T-flasks for transfection or plaque assays. |
| Minimum Split Density | Do not go below ( 3.0 \times 10^5 ) cells/mL [53] | Splitting at too low a density can stress the cells and lead to prolonged lag phases. |
| Maximum Split Density | Do not exceed ( 3.0 \times 10^6 ) cells/mL [53] | Allowing cells to become over-confluent can lead to reduced viability and nutrient depletion. |
| Target Passage Density | Split when density reaches ~( 2.0 \times 10^6 ) cells/mL [53] | This ensures cells are consistently passaged during their exponential growth phase. |
| Typical Split Ratio | 1:3 to 1:5 | The ratio depends on the desired passage schedule and growth rate; Sf9 may tolerate higher split ratios due to faster growth. |
General Subculture Protocol
Methodology for Passaging Adherent Sf9 and Sf21 Cells
- Preparation: Pre-warm complete culture medium (e.g., Sf-900 II SFM, EX-CELL 401, or IPL-41) to 28°C. Aseptically prepare the required number of new culture flasks [54].
- Cell Detachment:
- For firmly adherent cells, gently tap the flask to dislodge them.
- If cells remain attached, rinse the monolayer with a sterile, calcium- and magnesium-free solution (e.g., PBS) and use a gentle dissociation reagent if necessary. Avoid trypsin, as it is generally not required for insect cell dissociation.
- Cell Count and Viability Assessment: Take a sample of the cell suspension and perform a cell count using a hemocytometer or automated cell counter. Assess viability via Trypan Blue exclusion; healthy cultures should maintain viability above 95% [55].
- Seeding New Cultures: Calculate the volume of cell suspension needed to seed the new flasks at the recommended density of ( 3.0 \times 10^5 ) cells/mL for suspension or ( 5.0 \times 10^4 ) cells/cm² for adherent culture. Transfer the appropriate volume to the new flasks containing fresh, pre-warmed medium.
- Incubation: Cap the flasks securely and incubate at 28°C. No CO₂ control is required. For suspension cultures, maintain in an orbital shaker incubator at 110-125 rpm without baffles to ensure proper aeration and prevent cell clumping [53].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents and Materials for Insect Cell Culture
| Item | Function/Application | Example Products & Notes |
|---|---|---|
| Serum-Free Medium | Supports high-density growth; reduces contamination risk and simplifies downstream processing. | Sf-900 II SFM, EX-CELL 401 [54] [55]. The industry standard for recombinant protein production. |
| Shaker Flasks (Non-baffled) | Suspension culture vessel. | Polycarbonate Erlenmeyer flasks [55]. Non-baffled design is recommended to prevent excessive shear stress on cells [53]. |
| Orbital Shaker Incubator | Maintains constant temperature and agitation for suspension cultures. | Setpoint: 28°C; Agitation: 110-125 rpm [53] [55]. |
| Transfection Reagent | Introducing recombinant bacmid DNA into insect cells to generate baculovirus. | ExpiFectamine Sf, or PEI (Polyethylenimine) [56] [55]. |
| Cell Viability Assay | Quantifying cell health and, indirectly, viral titers. | MTT assay [55], or Trypan Blue exclusion for routine monitoring. |
Workflow for Subculturing and Application
The following diagram illustrates the critical decision points and pathways for the routine subculture of Sf9 and Sf21 cells, leading to their primary research applications.
Troubleshooting and Key Considerations
- Serum-Free Adaptation: Transitioning from serum-supplemented to serum-free medium must be gradual. A direct switch can cause a significant increase in doubling time. A recommended protocol is to transition over several passages using medium mixtures: begin with 75% old medium/25% new SFM, then 50:50, then 25:75, before finally moving to 100% SFM. This process can take 1-2 months but is critical for maintaining cell health [53].
- Cell Health Monitoring: Healthy, logarithmically growing Sf9 and Sf21 cells should have a doubling time of approximately 18-20 hours [53]. A prolonged doubling time indicates suboptimal conditions. Regularly monitor cell morphology and size; an increase in cell diameter is a key indicator of baculovirus infection [56].
- Antibiotic Use: Insect cells are sensitive to many chemicals. If antibiotics are necessary, consider using them at half-strength to avoid cytotoxic effects [53].
Preventing Cell Aggregation and Ensuring Single-Cell Suspensions for Reproducibility
In the field of adherent insect cell research, achieving and maintaining a high-quality single-cell suspension is a critical prerequisite for experimental reproducibility. Cellular aggregation poses a significant challenge, leading to inconsistent cell growth, variable metabolic activity, and irreproducible experimental outcomes across different research laboratories [57] [58]. The very nature of insect cells, which often attach strongly to substrates and to each other, compounds this problem, particularly under serum-free conditions commonly used in bioprocessing [1] [59].
This application note addresses the fundamental causes of cell clumping and provides detailed, actionable protocols to prevent aggregation. By integrating specific techniques such as surface modification, enzymatic treatment, and optimized handling practices, researchers can significantly improve the reliability of their insect cell cultures, thereby enhancing the validity of data generated in drug development and basic research applications.
Understanding the Causes of Cell Aggregation
Cell clumping arises from several interconnected factors, each requiring a distinct mitigation strategy. A comprehensive understanding of these mechanisms is the first step toward achieving reproducible single-cell suspensions.
Released DNA from Dying Cells: Physical stress during subcultivation, enzymatic dissociation, or freeze-thaw cycles can accelerate cell death. Dying cells release internal contents, including long strands of DNA, which act as a sticky glue that entangles neighboring cells and promotes the formation of large aggregates [60] [61]. This is a primary mechanism of clumping in stressed cultures.
Strong Cell-Surface Adherence: Insect cells, such as Sf9 cells, are known to adhere tenaciously to the hydrophilic surfaces of conventional glass culture vessels [58]. This uncontrolled adhesion not only results in considerable cell loss but also creates a visible cell rim, which can detach intermittently and form heterogeneous clumps within the suspension.
Enzymatic and Mechanical Stress: The process of dissociating adherent cells using enzymes like trypsin can itself be a source of stress. Over-digestion or the use of inappropriate enzymes can damage cell surface proteins, increasing the likelihood of aggregation as a stress response [62] [61].
Inadequate Culture Conditions: Allowing cultures to become over-confluent leads to increased cell lysis and the release of cellular debris. Furthermore, contamination with bacterial or fungal pathogens can directly cause cell death, exacerbating the clumping problem [61].
Core Strategies for Prevention and Intervention
Surface Modification of Culture Vessels
Silanizing glassware is a simple, cost-effective, and highly effective method for reducing cell adhesion and associated clumping, especially in small-scale cultivation.
Experimental Protocol: Silanization of Glassware Materials Required: Clean glass culture vessels, Sigmacote (or equivalent siliconeizing agent), distilled water, oven.
- Ensure glassware is clean, dry, and sterile.
- Fill the vessel with an adequate volume of Sigmacote (e.g., 3 mL for a 10 mL tube) [58].
- Swirl the vessel for 2 minutes to ensure complete coverage of the internal surface.
- Remove the silicone oil (which can be stored at 4°C for reuse).
- Rinse the vessel thoroughly six times with distilled water to remove any residual solution.
- Dry the glassware at 100°C for 1 hour.
- Sterilize in a drying oven at 180°C for 2 hours prior to use [58].
Impact of Silanization on Sf9 Cell Culture: A Quantitative Summary Table 1: The effect of vessel silanization on Sf9 cell growth parameters at different scales.
| Culture Vessel | Treatment | Lag Phase | Max Cell Density | Viability | Inter-replicate Variance |
|---|---|---|---|---|---|
| 10 mL Tube | Non-silanized | Present | Low | Lower | High |
| 10 mL Tube | Silanized | Absent | Tripled | Higher | Reduced |
| 50 mL Flask | Non-silanized | ~24 hours | Baseline | Baseline | Higher |
| 50 mL Flask | Silanized | ~0 hours | Slightly Increased | Slightly Higher | Reduced |
| 250 mL Flask | Non-silanized | Baseline | Baseline | Baseline | Baseline |
| 250 mL Flask | Silanized | Baseline | No Significant Change | No Significant Change | No Significant Change |
Data adapted from Scientific Reports (2024) [58]. The positive effects of silanization are most pronounced in small-scale vessels.
Enzymatic and Chemical Interventions
For clumps caused by released DNA or cation-dependent adhesion, specific additives can be incorporated into the culture and processing media.
Application Note: Using DNase I to Reduce Clumping Principle: DNase I enzymatically digests the free DNA released from dead cells, fragmenting the sticky mesh that holds cell aggregates together [60] [63]. Protocol:
- After thawing or dissociating cells, centrifuge and discard the supernatant.
- If the cell pellet appears clumpy, resuspend it in a pre-warmed culture medium or buffer containing DNase I at a final concentration of 100 µg/mL [60].
- Incubate the cell suspension at room temperature for 15 minutes.
- Wash the cells by adding medium with 2% FBS and centrifuging at 300 x g for 10 minutes to remove the DNase.
- Resuspend the pellet in fresh medium. If clumps persist, filter the suspension through a 37-70 µm cell strainer [60]. Note: DNase I should not be used if downstream applications involve DNA extraction. For RNA work, an RNase-free version is recommended [60].
Application Note: Using Chelators to Disrupt Cation-Dependent Clumping Principle: Cell adhesion in some cell types is dependent on cations like calcium and magnesium. EDTA, a cation chelator, can disrupt these interactions [63]. Protocol: Add 2 mM EDTA to wash buffers and media during cell preparation and staining phases. This is particularly effective for cells that clump via cation-dependent mechanisms [63].
Optimized Handling and Mechanical Dispersal
Proper physical handling is crucial to minimize stress and mechanically discourage aggregation.
- Gentle Resuspension: Avoid vigorous vortexing. For fragile cells, resuspend pellets by gentle flicking of the tube or slow, repetitive pipetting (trituration) with a wide-bore pipette tip [61] [63].
- Judicious Use of Pipetting: Trituration can break weak bonds between cells. Pass the suspension through a pipette 5-10 times gently before counting or subculturing.
- Filtration as a Last Resort: For persistent, small clumps, pass the entire cell suspension through a 70 µm cell strainer immediately before use. This ensures that only single cells and small, uniform clusters remain [60] [63].
- Proper Centrifugation: Use the correct speed and time (e.g., 200-300 x g for 5-10 minutes) to pellet cells without promoting tight packing that leads to clumping [1] [61].
Integrated Workflow for a Perfect Single-Cell Suspension
The following diagram synthesizes the core strategies into a logical workflow for preventing and addressing cell aggregation, from preparation to final quality control.
Integrated Workflow for Single-Cell Suspension
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key research reagent solutions for preventing cell aggregation.
| Reagent/Material | Function & Mechanism of Action | Example Application/Note |
|---|---|---|
| Sigmacote | Silanizing agent that creates a hydrophobic, chemically inert layer on glass, reducing cell adhesion. | Cost-effective; coating is durable and can be renewed. Ideal for small-scale glass vessels [58]. |
| DNase I | Endonuclease that fragments free DNA released by dead cells, dissolving the "glue" that causes clumping. | Use at 100 µg/mL for 15 min incubation. Do not use for downstream DNA extraction [60] [63]. |
| EDTA | Chelating agent that binds divalent cations (Ca²⁺, Mg²⁺), disrupting cation-dependent cell adhesion. | Add at 2 mM to buffers and media. Avoid if studying cation-dependent cell functions [63]. |
| TrypLE / Accutase | Gentle, enzyme-based cell detachment solutions. Cleave cell-surface attachments without degrading many cell surface proteins like trypsin. | Safer alternative to trypsin for generating single-cell suspensions for flow cytometry and subculturing [62] [63]. |
| Cell Strainers | Physical removal of large, persistent clumps via size exclusion, ensuring a uniform suspension. | Use 70 µm strainers prior to staining or analysis to prevent instrument blockages and ensure even staining [60] [63]. |
| Polypropylene Tubes | Low-binding plastic material that minimizes cell attachment compared to standard polystyrene. | Use for storing and processing cell suspensions to minimize cell loss on container walls [63]. |
Concluding Remarks
Achieving reproducibility in adherent insect cell research is fundamentally linked to the quality of the single-cell suspension. By understanding the root causes of aggregation—released DNA, surface adhesion, and handling stress—researchers can deploy a targeted toolkit to mitigate the problem. The integrated workflow combining preventive vessel silanization, judicious use of enzymatic helpers like DNase I, and optimized mechanical handling provides a robust framework for significantly improving experimental consistency. For the drug development professional, these protocols are not merely suggestions but essential components of a rigorous and reliable cell culture process, ensuring that data generated from insect cell systems is both trustworthy and reproducible.
Benchmarking Success: Validation Methods and Cross-System Comparisons
Within the broader scope of thesis research on subculturing adherent insect cells, the rigorous monitoring of key performance indicators (KPIs) is fundamental to experimental reproducibility and scalability. For researchers and drug development professionals, the insect cell-baculovirus expression vector system (BEVS) has become an indispensable platform for the production of complex biologics, including vaccines, viral vectors, and therapeutic proteins [13]. The global insect cell lines market, projected to grow from USD 1,245.4 Million in 2025 to USD 3,665.8 Million by 2035, underscores the critical importance of robust and standardized cell culture protocols [64].
Effective process control hinges on the accurate assessment of cellular health and growth kinetics. This Application Note details the essential protocols for quantifying three core KPIs—viability, doubling time, and morphology—in adherent insect cell cultures, such as the widely used Sf9 and High Five lines. Mastering these techniques ensures optimal cell growth, maximizes recombinant protein yields, and provides early detection of culture abnormalities, thereby de-risking biopharmaceutical development pipelines.
Key Performance Indicators (KPIs) for Adherent Insect Cells
The following table summarizes the three critical KPIs, their biological significance, and the methodologies for their assessment.
Table 1: Core Key Performance Indicators for Adherent Insect Cell Culture
| Key Performance Indicator (KPI) | Purpose & Significance | Measurement Method | Target Range for Healthy Cultures |
|---|---|---|---|
| Cell Viability | Assesses the percentage of living cells in a population; indicates culture health and suitability for subculturing or infection. | Trypan Blue Exclusion Assay [65] | >90% at the time of subculturing [65] |
| Population Doubling Time | Measures the rate of cell proliferation; essential for determining split ratios and scheduling production runs. | Calculation from cell concentration data over exponential growth phase. | Varies by cell line, but consistent, low doubling times indicate health. Increases signal stress [65]. |
| Cell Morphology | A macroscopic indicator of cellular physiology; changes can signal infection, stress, or adaptation to culture conditions. | Phase Contrast Microscopy & Image Analysis (e.g., UWT-MIA) [66] | Uniform, adherent cells specific to the line (e.g., Sf9). Deviations indicate unhealthy culture [65]. |
Experimental Protocols
Protocol 1: Passaging Adherent Insect Cells
This protocol describes the subculturing of adherent insect cells, a critical process during which KPIs are routinely monitored.
Principle: Adherent insect cells require passaging to maintain logarithmic growth. This involves detaching cells from the substrate using enzymatic and mechanical means, then diluting them into fresh culture vessels [65].
Materials:
- Adherent insect cell culture (e.g., Sf9, Sf21)
- Insect cell-specific culture medium (e.g., Grace's Medium) [65]
- Dissociation reagent (e.g., Trypsin or TrypLE) [65]
- Balanced salt solution without calcium and magnesium (e.g., PBS)
- Complete growth medium
- Centrifuge tubes
- Hemocytometer or automated cell counter (e.g., Invitrogen Countess) [65]
- Trypan Blue solution (0.4%)
Procedure:
- Monitor Culture: Routinely observe cells under a phase-contrast microscope. Passage cells during the log phase, before they reach confluency, unless they are strongly adherent. For strongly adherent lines, passage at confluency or slightly after when cells begin to pull away. Avoid passaging at densities below 20% confluency or repeatedly after confluency, as both practices decrease doubling times and viability [65].
- Remove Medium: Aspirate and discard the spent cell culture medium from the culture vessel.
- Wash Cell Layer: Gently add a balanced salt solution without calcium and magnesium to the side of the vessel opposite the cell layer. Rock the vessel to wash the monolayer, then remove and discard the solution. This step removes serum and ions that inhibit dissociation reagents [65].
- Add Dissociation Reagent: Add pre-warmed dissociation reagent to the side of the flask, using enough to cover the cell layer (e.g., ~0.5 mL per 10 cm²) [65].
- Incubate and Observe: Incubate the vessel at room temperature for approximately 2 minutes. Actual incubation time varies by cell line. Observe under a microscope for cell detachment. If less than 90% of cells are detached, tap the vessel and/or continue incubation, checking every 30 seconds [65].
- Neutralize Reaction: When ≥90% of cells are detached, add a volume of pre-warmed complete growth medium equivalent to twice the volume of the dissociation reagent. Disperse the medium by pipetting over the cell layer surface to ensure complete cell detachment and to neutralize the enzyme [65].
- Transfer and Centrifuge: Transfer the cell suspension to a centrifuge tube and pellet the cells at approximately 200 x g for 5-10 minutes [65].
- Resuspend and Count: Resuspend the cell pellet in a small volume of fresh medium. Take a sample for cell counting and viability assessment using the Trypan Blue exclusion method (detailed in Protocol 2).
- Seed New Flasks: Dilute the cell suspension to the recommended seeding density and pipet into new culture vessels.
- Return to Incubator: Culture insect cells at 27°C in a non-humidified environment. CO₂ exchange is not typically required [65].
Protocol 2: Determining Viability and Doubling Time
Principle: Viability is determined by a dye exclusion assay, where non-viable cells with compromised membranes take up Trypan Blue. Doubling time is calculated from the increase in viable cell concentration over time.
Materials:
- Cell suspension
- Trypan Blue solution (0.4%)
- Hemocytometer or automated cell counter
- Microcentrifuge tubes
Procedure:
- Prepare Cell Sample: Ensure the cell suspension is well-mixed. For adherent cells, follow the passaging protocol (Protocol 1) to obtain a single-cell suspension.
- Mix with Trypan Blue: Mix 10 μL of cell suspension with 10 μL of 0.4% Trypan Blue solution. Allow to incubate for 1-2 minutes. Do not exceed 5 minutes.
- Load Hemocytometer: Carefully load the mixture into both chambers of the hemocytometer.
- Count Cells: Under a microscope, count the total number of cells and the number of blue-stained (non-viable) cells in the four large corner squares of the grid.
- Calculate Concentration and Viability:
- Total Cell Concentration (cells/mL) = (Total cells counted / 4) × Dilution Factor × 10⁴
- Viable Cell Concentration (cells/mL) = (Viable cells counted / 4) × Dilution Factor × 10⁴
- Percent Viability = (Viable Cell Concentration / Total Cell Concentration) × 100%
- Dilution Factor = 2 (if using equal volumes of cell suspension and Trypan Blue)
- Calculate Population Doubling Time:
- Measure the viable cell concentration at two time points during the exponential growth phase (t₁ and t₂).
- Doubling Time (T_d) = (t₂ - t₁) × ln(2) / ln(VC₂ / VC₁)
- Where VC₁ and VC₂ are the viable cell concentrations at times t₁ and t₂, respectively.
Protocol 3: Assessing Cell Morphology via Image Analysis
Principle: Phase contrast microscopy (PCM) allows for non-invasive, long-term observation of live cells. Advanced image analysis, such as Undecimated Wavelet Transform Multivariate Image Analysis (UWT-MIA), can extract textural features from PCM images that are quantitatively correlated with cellular morphology [66].
Materials:
- Phase contrast microscope with a time-lapse imaging system (optional)
- Live cell culture in an appropriate imaging vessel
- Image analysis software (e.g., with UWT-MIA capability)
Procedure:
- Image Acquisition: Place the culture vessel on the microscope stage. Acquire phase contrast images of the cell monolayer at regular intervals (e.g., every 4-8 hours) over the culture period. Ensure consistent lighting and focus.
- Feature Extraction (UWT-MIA):
- Apply an undecimated wavelet transform to the acquired images to decompose them into different frequency bands.
- From these wavelet coefficients, compute textural features that describe the pattern and structure of the cell monolayer.
- Model Building: Establish a correlation between the extracted textural features and specific morphological parameters (e.g., major/minor axis length, roundness) using a partial least squares (PLS) regression model. This requires a "ground truth" set of images where the morphological features have been manually quantified [66].
- Morphology Monitoring: Apply the trained PLS model to new, incoming live-cell images. The model can then predict the morphological state of the culture in real-time, allowing for the detection of subtle changes due to growth, media conditions, or other stressors [66].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Adherent Insect Cell Culture Experiments
| Item | Function & Application |
|---|---|
| Sf9 Cell Line | A clonal isolate of Spodoptera frugiperda pupal ovarian tissue; the industry standard for baculovirus-mediated recombinant protein expression due to high transfection efficiency and scalability [64]. |
| Grace's Insect Medium | A specially formulated, slightly acidic growth medium designed to support the growth of a wide variety of insect cells [65]. |
| Serum-Free Insect Cell Media | Chemically defined formulations that eliminate batch-to-batch variability, enhance reproducibility, and reduce contamination risk for biopharmaceutical manufacturing [34] [67]. |
| TrypLE Express | A recombinant trypsin-like enzyme for cell dissociation; a stable, ready-to-use alternative to purified trypsin that minimizes variability and supports standardized downstream processes [65]. |
| Cytodex 1 Microcarriers | Dextran-based microcarriers providing a high surface-to-volume ratio for scaling up adherent insect cell culture in stirred-tank bioreactors [68]. |
Workflow for KPI Monitoring
The following diagram illustrates the integrated workflow for monitoring the health of adherent insect cell cultures, from subculturing to data-driven decision-making.
Concluding Remarks
The systematic monitoring of viability, doubling time, and morphology provides a powerful framework for ensuring the health and productivity of adherent insect cell cultures. Integrating these KPIs into routine subculturing protocols, as part of a comprehensive thesis investigation, builds a foundation of process understanding and control. As the biopharmaceutical industry continues to leverage the insect cell-BEVS platform for next-generation vaccines and therapeutics [13], these foundational techniques will remain paramount in translating research from the bench to scalable, robust manufacturing processes.
How Insect Cell Subculture Differs from Mammalian Cell Culture Protocols
Within the context of broader research on subculturing adherent insect cells, it is essential to recognize that insect and mammalian cell cultures constitute fundamentally distinct biological systems, each demanding specific handling procedures. These differences stem from their unique evolutionary origins, physiological requirements, and cellular characteristics. Mammalian cells, typically derived from human or other mammalian tissues, require conditions that closely mimic their native physiological environment [69]. In contrast, insect cells, commonly derived from lepidopteran species such as Spodoptera frugiperda, exhibit greater environmental flexibility and possess different cellular machinery [14] [17]. Understanding these distinctions is critical for researchers, particularly in drug development and recombinant protein production, where optimal cell health and productivity are paramount.
The most commonly used insect cell lines, Sf9 and Sf21, originate from the pupal ovarian tissue of the fall armyworm (Spodoptera frugiperda) [70] [17]. These are frequently employed with the baculovirus expression vector system (BEVS) for recombinant protein production [14] [17]. In mammalian cell culture, HEK-293T cells serve as a workhorse for viral production and protein expression [70]. These cellular origins dictate vastly different requirements for temperature, gas exchange, nutrition, and physical handling, which form the basis for the protocol variations detailed in this application note.
Core Differences in Culture Conditions and Handling
The table below summarizes the fundamental environmental and handling differences between insect and mammalian cell culture systems, which must be rigorously observed to maintain cell viability and experimental reproducibility.
| Parameter | Insect Cells | Mammalian Cells |
|---|---|---|
| Incubation Temperature | 27–28°C [1] [59] | 37°C [1] [59] |
| CO₂ Requirement | Not required [1] [59] | 5% CO₂ typically required [1] [59] |
| Buffer System | Phosphate buffer [59] | Bicarbonate/HEPES buffer [59] |
| Humidity Control | Non-humidified environment sufficient [1] | ~95% relative humidity typically required [59] |
| Growth Characteristics | Can be strongly adherent; some lines grow in suspension [17] | Most primary lines are strictly adherent; some transformed lines grow in suspension [4] [45] |
| Doubling Time | Generally fast-growing [69] | Generally slower-growing [69] |
| Subculturing Confluence | Passage at confluency for strongly adherent lines [1] | Passage at log phase, before confluency [71] |
Special Considerations for Subculturing
Insect Cells: Strongly adherent insect cell lines, such as Sf9 and Sf21, require special techniques for detachment. Unlike mammalian cells, they attach very tightly to substrates under serum-free conditions and often need mechanical force for dislodging [1]. The recommended method is to strike the flask sharply with the palm of the hand 2-3 times after a brief refrigeration period [5]. Enzymatic detachment with trypsin is less commonly required than in mammalian culture.
Mammalian Cells: Standard subculturing involves enzymatic digestion (e.g., trypsin or TrypLE) to break down cell-surface proteins after a wash step with a balanced salt solution to remove calcium and magnesium, which inhibit trypsin activity [1]. The process is typically faster and requires less physical manipulation.
Detailed Experimental Protocols
Protocol for Subculturing Adherent Mammalian Cells
Principle: Mammalian cells require enzymatic release from the culture surface using trypsin or similar proteases after removal of inhibitory ions, followed by dilution into new culture vessels to maintain logarithmic growth [1] [71].
Materials:
- Pre-warmed complete growth medium
- Balanced salt solution without calcium and magnesium (e.g., PBS)
- Pre-warmed dissociation reagent (e.g., trypsin or TrypLE)
- Centrifuge tubes
- Hemocytometer or automated cell counter
- Trypan blue solution (0.4%)
Method:
- Preparation: Monitor cell viability and confluence. Proceed when cells are in the log phase (typically 70-90% confluence) with viability >90% [71].
- Wash: Remove and discard spent media. Wash the cell layer with a balanced salt solution without calcium and magnesium (approx. 2 mL per 10 cm²) to remove serum residues and inhibitory ions [1].
- Detach: Add pre-warmed dissociation reagent (approx. 0.5 mL per 10 cm²) to cover the cell layer. Incubate at room temperature for approximately 2 minutes (times vary by cell line) [1].
- Confirm Detachment: Observe under a microscope. If <90% of cells are detached, extend incubation in 30-second increments with gentle tapping [1].
- Neutralize: When most cells are detached, add 2 volumes of complete growth medium to neutralize the enzyme. Pipette gently to disperse cells [1].
- Centrifuge: Transfer cell suspension to a centrifuge tube. Centrifuge at 200 × g for 5-10 minutes. Discard supernatant [1].
- Resuspend and Count: Resuspend cell pellet in a small volume of fresh medium. Remove a sample for counting with a hemocytometer using Trypan blue exclusion to determine viability and total cell count [1] [71].
- Seed: Dilute cell suspension to the recommended seeding density (varies by cell line) and transfer to new culture vessels. Loosen caps for gas exchange and incubate at 37°C with 5% CO₂ [1] [45].
Protocol for Subculturing Adherent Insect Cells (Sf9/Sf21)
Principle: Insect cells often require mechanical dislodgment due to strong adherence, and are cultured at lower temperatures without CO₂ control in specialized media [1] [5] [17].
Materials:
- Insect cell culture medium (e.g., Grace's medium, TNM-FH, SF-900 II) [5] [17]
- 25-cm² tissue culture flasks
- Sterile pipettes
- 70% ethanol
- Refrigerator (4°C)
Method:
- Determine Confluence: Passage strongly adherent insect cells like Sf9 and Sf21 at confluency or slightly after, when they may start to pull away from the flask bottom. Passaging before confluency requires more force and can decrease cell health over time [1].
- Cool: Place the mature culture in a refrigerator (4°C) for 20 minutes. Cooling helps weaken cell attachment [5].
- Mechanical Dislodgment: Hold the culture flask firmly and strike it sharply with the palm of the other hand 2-3 times. Avoid vigorous shaking to prevent cell damage [1] [5].
- Visual Inspection: Observe under a microscope to confirm detachment. If cells remain attached, repeat the striking process.
- Transfer: Loosen the cap and use a sterile pipette to transfer the appropriate volume of cell suspension into a new flask containing fresh, pre-warmed medium [5]. A 1:10 split ratio is common, but optimal ratios vary by cell line and application [5].
- Incubate: Tighten caps and incubate cultures at 27°C in a non-humidified, non-CO₂ environment. A controlled 27°C incubator is recommended, though cultures can be maintained on benchtops if protected from light [1].
The Scientist's Toolkit: Essential Research Reagents
The table below lists key materials required for successful insect and mammalian cell culture, based on protocols from leading suppliers and research institutions.
| Reagent/Equipment | Function | Insect Cell Specifics | Mammalian Cell Specifics |
|---|---|---|---|
| Basal Medium | Provides nutrients, salts, and pH buffer | Grace's Medium, TNM-FH [5] [17]; more acidic (pH 6.0-6.8) [59] | DMEM, RPMI-1640; higher pH (7.0-7.3) [59] |
| Serum/Supplement | Supplies growth factors and adhesion factors | Often supplemented with fetal bovine serum (FBS) or growth-promoting substances [59] | Typically requires 5-10% FBS [1] |
| Dissociation Agent | Releases adherent cells from substrate | Primarily mechanical dislodgement [1] [5] | Trypsin or TrypLE [1] |
| Incubator | Maintains optimal growth environment | 27°C, non-humidified, ambient CO₂ [1] [59] | 37°C, ~95% humidity, 5% CO₂ [1] [59] |
| Cryoprotectant | Prevents ice crystal formation during freezing | DMSO (5-10%) or Glycerol (2-20%) in serum [4] | Typically DMSO (5-10%) in serum or serum-containing media [4] |
Troubleshooting Common Cell Culture Problems
Successful cell culture requires diligent monitoring and rapid response to issues. The table below outlines common problems, their causes, and solutions specific to insect and mammalian cell systems.
| Problem | Possible Cause | Suggested Solution |
|---|---|---|
| Poor Cell Attachment (Insect) | Sub-optimal confluence at passaging | For strongly adherent lines, passage at confluency, not before [1] |
| Difficulty Detaching (Insect) | Excessive adherence under serum-free conditions | Use quick, wrist-snapping mechanical shock; ensure cells are at appropriate confluence [1] |
| Rapid pH Drop | Buildup of lactic acid from high cell metabolism | Subculture cells before they reach stationary phase; replace medium more frequently [71] |
| Decreased Doubling Time | Repeated passaging at incorrect density | For insect cells, avoid passaging both before confluency and long after confluency [1] |
| Contamination | Break in aseptic technique | Use antibiotics if appropriate; maintain strict sterile technique; aliquot reagents [45] |
Mastering the distinct protocols for insect and mammalian cell subculturing is fundamental to research success in biopharmaceutical development. The differences in their biological origins—insects being ectothermic invertebrates and mammals being endothermic vertebrates—directly translate to specific technical requirements for temperature, gas exchange, and physical handling [69] [59]. Adherence to these specialized protocols ensures the health of the cell lines, the reproducibility of experiments, and the reliability of downstream products, whether they be recombinant proteins, viruses, or other biologics.
For researchers framing their work within a broader thesis on adherent insect cell research, recognizing these distinctions is not merely procedural but conceptual. The robustness and efficiency of insect cell systems, particularly the Sf9 and Sf21 lines used with BEVS, offer significant advantages for scalable protein production [14] [17]. However, these advantages are only fully realized through meticulous attention to their unique culturing demands, which differ profoundly from the more familiar mammalian cell culture paradigms.
The cultivation of adherent insect cells presents a significant challenge in bioprocessing, particularly when transitioning from laboratory-scale research to industrial-scale production. The hybrid approach, utilizing microcarriers, bridges this gap by providing a high-surface-area substrate within a suspension culture environment. This system is especially relevant for entomoculture, the cultivation of insect cells for food and bioproducts, where scalable and cost-effective production is paramount [72]. Unlike traditional static flasks, microcarrier-based cultures in stirred-tank bioreactors enable efficient gas exchange, nutrient distribution, and environmental control, supporting cell densities that far exceed conventional methods [33].
Insect cells, such as the Manduca sexta non-adherent cells (MsNACs) described in recent cultivated meat research, demonstrate remarkable adaptability to suspension culture and can achieve densities exceeding 20 million cells per mL in optimized shake flask cultures [72]. This robust growth potential, combined with the ability to thrive in animal component-free media, positions insect cells as an ideal candidate for microcarrier-based expansion systems. Furthermore, insect cells offer practical advantages including room-temperature growth without CO₂ supplementation, significantly simplifying incubation systems and reducing facility costs compared to mammalian cell culture [72].
Key Experimental Protocols
Microcarrier Preparation and Cell Seeding
Proper preparation of microcarriers is essential for successful cell attachment and growth. The following protocol outlines the key steps for initiating a microcarrier-based insect cell culture.
Microcarrier Preparation: Hydrate and sterilize microcarriers (e.g., Cytodex 1 or cellulose-based DE-53) according to manufacturer specifications. Typically, this involves swelling in calcium- and magnesium-free phosphate-buffered saline (PBS) followed by autoclaving or gamma irradiation. For charge-modified microcarriers, ensure complete equilibration with the culture medium to establish proper surface charge for cell attachment [73] [74].
Cell Seeding Optimization: Dissociate adherent insect cells from conventional culture vessels using a plant-origin dispersion agent (RDB) or trypsin-EDTA, as insect cells can attach very tightly under serum-free conditions [1] [73]. Determine viable cell density using trypan blue exclusion and automated cell counting. Seed cells at an optimized density of >16 cells per microcarrier bead onto 3.5–4.5 g/L of prepared microcarriers in a stirred-tank bioreactor [75]. Initiate agitation at the minimum speed required to maintain suspension (Njs) to facilitate initial cell attachment without causing excessive shear stress [75].
Culture Maintenance and Monitoring
Once cells are successfully seeded, maintaining optimal culture conditions is critical for achieving high cell densities.
Agitation and Feeding Strategy: Maintain insect cells at 27°C in a non-humidified environment with continuous, gentle agitation. After the first 24 hours, agitation speed may be gradually increased by a maximum of 25% to improve mixing as cell density increases [1] [75]. Implement a feeding strategy based on nutrient consumption rates; for example, replace 50% of the spent medium with fresh, rich medium every 2-3 days to maintain nutrient levels and remove metabolic wastes [75].
Process Monitoring and Harvesting: Monitor key parameters including dissolved oxygen, pH, and glucose consumption regularly. Observe cell growth and confluence on microcarriers through periodic sampling and microscopic examination. For harvesting, perform in-situ detachment using trypsin-EDTA solution (approximately 90 mL per gram of microcarriers) with agitation at 100 rpm for 25-30 minutes [75]. Confirm complete cell detachment microscopically before proceeding to the next passage or downstream application.
Scale-Up Strategy via Bead-to-Bead Transfer
For efficient scale-up without the need for additional static culture, implement a bead-to-bead transfer protocol during subculturing.
In-Situ Detachment and Transfer: After achieving target confluency on microcarriers, perform in-situ cell detachment as described above. Instead of separating cells from spent microcarriers, transfer the entire mixture of detached cells and spent microcarriers directly to a new bioreactor containing fresh medium and additional microcarriers at a split ratio of 1:5 [75]. This approach, known as "bead-to-bead transfer," enhances cell recovery and reduces process complexity by eliminating the need for microcarrier separation steps [75].
Serial Subculturing Validation: Validate the bead-to-bead transfer process through serial subculturing. Research demonstrates that MA 104 cells can be successfully subcultured for at least five passages on Cytodex 1 microcarriers using this method without significant loss of viability or growth potential [75]. This approach streamlines the seed train process and facilitates direct scale-up from laboratory to production scales.
Experimental Data and Analysis
Quantitative Performance of Culture Systems
The following table summarizes key quantitative data from microcarrier-based culture systems, highlighting the enhanced performance achievable with this technology.
Table 1: Performance Metrics of Microcarrier-Based Culture Systems
| Parameter | Traditional 2D Culture | Microcarrier-Based Culture | Data Source |
|---|---|---|---|
| Maximum Cell Density (cells/mL) | Limited by surface area | 2.6 × 10⁶ (MA 104 on Cytodex 1) [75] | [75] |
| Surface Area to Volume Ratio (cm²/mL) | ~0.5-5 (T-flasks) | 120-160 [76] | [76] |
| Scale-Up Potential | Scale-out required (multiple vessels) | Linear scale-up to >1000L [74] | [74] |
| Relative Labor Intensity | High (manual handling) | Moderate (automated systems) [74] | [74] |
| Harvest Efficiency | Variable | >93% with automated systems [76] | [76] |
Microcarrier Selection Guide
Selecting the appropriate microcarrier is critical for optimizing insect cell growth. The table below compares different microcarrier types and their applications.
Table 2: Microcarrier Types and Their Applications in Insect Cell Culture
| Microcarrier Type | Material/Coating | Key Features | Compatible Cell Lines |
|---|---|---|---|
| Cytodex 1 | DEAE-dextran | Charge-modified; widely used | MA 104, Sf9, High-Five [75] |
| Cellulose-based (DE-53) | Microgranular cellulose | Ion-exchange capacity; supports serum-free culture | Aedes aegypti (AA), mosquito cell lines [73] |
| BioNOC II | Polyethylene Terephthalate (PET) | Macroporous; 3D growth; high surface area (2,400 cm²/g) | Sf-9, Hi-5 [76] |
| Collagen-coated | Polystyrene with collagen | Enhanced cell attachment; biocompatible | Sensitive insect cell lines [74] |
Research Reagent Solutions
The following table outlines essential materials and reagents required for establishing and maintaining microcarrier-based insect cell cultures.
Table 3: Essential Research Reagents for Microcarrier-Based Insect Cell Culture
| Reagent/Carrier | Function | Application Notes |
|---|---|---|
| Cytodex 1 Microcarriers | Provides surface for cell attachment | Use at 3.5-4.5 g/L concentration; optimal for bead-to-bead transfer [75] |
| SF900 III Medium | Serum-free growth medium | Supports high-density insect cell growth; animal component-free [72] |
| Trypsin-EDTA (0.25%) | Cell dissociation from microcarriers | Use 90 mL/g MC for in-situ detachment; neutralization with serum required [75] |
| RDB Dispersion Agent | Plant-based cell dissociation | Alternative to trypsin; effective for tightly adherent insect cells [73] |
| BioNOC II Macrocarriers | Macroporous carrier for 3D growth | Suitable for packed-bed bioreactors; high surface area [76] |
Technical Considerations and Workflow
Process Optimization Parameters
Successful implementation of microcarrier technology requires careful optimization of several critical process parameters.
Agitation Control: Implement a controlled agitation regimen starting at the minimum speed required for suspension (Njs) to facilitate initial cell attachment during the first 24 hours. Gradually increase speed thereafter, not exceeding 25% above Njs, to maintain homogeneity while minimizing shear stress that can damage cells or detach them from microcarriers [75]. The precise agitation rate must balance adequate mixing with the shear sensitivity of the specific insect cell line being cultured.
Inoculation Density and Microcarrier Concentration: Optimize the cell-to-bead ratio to achieve a seeding density of >16 cells/bead, which promotes rapid colonization and minimizes empty microcarriers. Use a microcarrier concentration of 3.5-4.5 g/L in the culture vessel to provide sufficient surface area while avoiding bead overcrowding that could lead to aggregation or nutrient gradients [75]. These parameters significantly impact the final cell yield and should be established for each cell line through design of experiments (DoE) approaches.
Decision Pathway for Culture Establishment
The following diagram illustrates the key decision points and workflow for establishing a successful microcarrier-based insect cell culture system.
Comparative Analysis of Culture Technologies
When selecting a culture platform, researchers should consider the relative advantages and limitations of available technologies.
Table 4: Technology Comparison for Scalable Adherent Culture
| Technology | Relative Surface Area | Scalability | Shear Stress | Process Control |
|---|---|---|---|---|
| T-Flasks/Multi-layers | Low | Limited (scale-out) | None | Low [33] |
| Roller Bottles | Moderate | Limited (scale-out) | Low | Low [33] |
| Microcarriers (Stirred Tank) | High | High (scale-up) | Moderate-High | High [33] [74] |
| Macroporous Carriers (Fixed-Bed) | Very High | Moderate | Very Low | Moderate [76] [33] |
The hybrid approach of utilizing microcarriers for scalable adherent insect cell culture represents a transformative methodology that effectively bridges the gap between laboratory research and commercial-scale production. By providing a high-surface-area environment within controlled bioreactor systems, this technology enables significant increases in cell yield while reducing labor requirements and footprint compared to traditional 2D culture systems [74]. The adaptability of insect cells to suspension culture on microcarriers, combined with their ability to thrive in animal component-free media, positions this platform as particularly valuable for emerging applications such as entomoculture for cultivated meat [72].
Future developments in microcarrier technology will likely focus on creating specialized surfaces tailored to insect cell attachment, implementing fully automated closed-system bioreactors, and designing biodegradable carriers that simplify downstream processing [74]. As the field of insect cell bioprocessing continues to evolve, the hybrid microcarrier approach will play an increasingly important role in enabling the efficient, scalable production of high-value biologicals, vaccines, and sustainable protein sources.
The insect cell expression system, particularly the baculovirus expression vector system (BEVS), has emerged as a powerful and versatile platform for producing complex biologics. This application note details its successful implementation in the production of vaccines and therapeutic proteins, framed within the context of subculturing and maintaining adherent insect cells. The platform's value was globally recognized during the COVID-19 pandemic, with several authorized vaccines originating from this technology [13]. Its ability to perform eukaryotic post-translational modifications while offering the scalability and cost-effectiveness of a non-mammalian system makes it an ideal choice for both research and industrial-scale production [14] [22]. This document provides a detailed overview of key commercial successes, supplemented with standardized protocols for cell culture and the essential reagents required to establish this system in a research laboratory.
Successful Commercial Applications
The insect cell-BEVS platform has transitioned from a research tool to a validated industrial manufacturing platform for human and veterinary medicines. The table below summarizes several key approved products.
Table 1: Approved Vaccines and Therapeutics Produced Using Insect Cell-BEVS
| Product Name | Application / Target | Antigen / Protein | Manufacturer/Sponsor | Approval Status & Year |
|---|---|---|---|---|
| Cervarix | Human Papillomavirus (HPV) | HPV16/18 L1 protein (VLP) | GSK | FDA Approved (2007) [13] |
| Provenge | Prostate Cancer | PAP-GM-CSF fusion protein | Dendreon | FDA Approved (2011) [13] |
| FluBlok/Flublok Quadrivalent | Influenza | Hemagglutinin (HA) protein | Sanofi Pasteur | FDA Approved (2013/2016) [13] |
| NVX-CoV2373 | COVID-19 | SARS-CoV-2 Spike (S) protein | Novavax | FDA Emergency Use Authorization (2020) [13] |
| Porcilis Pesti | Classical Swine Fever | E2 protein | MSD Animal Health | Approved (2000) [13] |
| CircoFLEX | Porcine Circovirus-2 | PCV2 ORF2 protein | Boehringer Ingelheim | Approved (2005) [13] |
Case Study: Novavax NVX-CoV2373 COVID-19 Vaccine
The NVX-CoV2373 vaccine is a recombinant nanoparticle vaccine produced using the BEVS platform in Sf9 insect cells derived from the fall armyworm, Spodoptera frugiperda [13]. The vaccine is based on the stable, pre-fusion conformation of the SARS-CoV-2 spike glycoprotein, which is engineered to mimic the native viral antigen. The use of the BEVS allowed for the rapid production and scaling of this recombinant protein. In Phase III clinical trials, the vaccine demonstrated an efficacy rate of 89.7%, highlighting the capability of the insect cell system to produce a highly immunogenic and protective antigen [13]. The success of NVX-CoV2373 underscores the platform's utility in responding swiftly to emerging pandemic threats with a safe and effective subunit vaccine.
Case Study: Cervarix HPV Vaccine
Cervarix was the first major virus-like particle (VLP)-based vaccine produced in insect cells to gain widespread approval. VLPs mimic the structure of the native virus but lack the viral genetic material, making them non-infectious and highly safe. Cervarix is produced by expressing the L1 major capsid protein of HPV types 16 and 18 in insect cells using BEVS [13]. These proteins self-assemble into VLPs that are structurally identical to the authentic virion, thereby eliciting a potent and protective immune response. The approval of Cervarix validated the insect cell platform for its ability to correctly fold and assemble complex, multimeric protein structures that are critical for vaccine efficacy.
Insect Cell Culture and Maintenance Protocols
The reliability of the insect cell expression system is fundamentally dependent on proper cell culture techniques. Below are the standard protocols for subculturing adherent insect cells and generating a recombinant baculovirus.
Protocol: Subculturing Adherent Insect Cells
This protocol is adapted for commonly used lepidopteran cell lines such as Sf9 and Sf21. Note: Unlike mammalian cells, insect cells are maintained at 27°C without CO~2~ supplementation and typically do not require a humidified environment [77] [22].
Table 2: Standard Culture Conditions for Common Insect Cell Lines
| Cell Line | Origin | Optimal Temperature | Common Culture Media | Key Features |
|---|---|---|---|---|
| Sf9 | Spodoptera frugiperda (ovarian) | 27°C ± 1°C | Sf-900 II/III SFM, Grace's Supplemented | High growth rate, tolerant to high densities and shear stress, ideal for virus amplification and protein production [22] [17] |
| Sf21 | Spodoptera frugiperda (ovarian) | 27°C ± 1°C | Sf-900 II/III SFM, Grace's Supplemented | Highly susceptible to viral infection, excellent for plaque assays [17] |
| High Five | Trichoplusia ni (ovarian) | 27°C ± 1°C | Express Five SFM | High yield of secreted proteins, but may produce more proteases [22] [17] |
Procedure:
- Monitoring: Routinely monitor cell health and confluency. Passage cells during the log phase, typically at 80-90% confluency, to maintain viability and prevent decline in health parameters [77] [17].
- Washing: Aspirate and discard the spent culture medium. Wash the cell monolayer with a balanced salt solution without calcium and magnesium (e.g., PBS) to remove traces of serum and ions that can inhibit dissociation agents. Use approximately 2 mL per 10 cm² surface area [77].
- Dissociation: Add a pre-warmed dissociation reagent, such as trypsin or a non-animal alternative like TrypLE (approximately 0.5 mL per 10 cm²). Gently rock the vessel to ensure complete coverage [77].
- Incubation & Detachment: Incubate the vessel at room temperature for 2-5 minutes. Observe cells under a microscope. For strongly adherent insect cells, a quick, sharp tap of the flask may be necessary to dislodge them. Avoid vigorous shaking to prevent cell damage [77].
- Neutralization: When ≥ 90% of cells have detached, add a volume of pre-warmed complete growth medium that is at least twice the volume of the dissociation reagent to neutralize its activity [77].
- Seeding: Gently pipette the cell suspension to break up clumps and seed appropriate volumes into new culture vessels containing fresh pre-warmed medium. Return the cells to the 27°C incubator [77].
Protocol: Recombinant Protein Production using BEVS
The Bac-to-Bac system is a widely used method for generating recombinant baculovirus. The workflow involves creating a recombinant bacmid in bacteria, followed by virus generation and protein expression in insect cells.
Key Steps Elaboration:
- Recombinant Bacmid Generation (Steps 1-4): The gene of interest is first cloned into a donor plasmid. This plasmid is then transformed into specialized E. coli cells (e.g., DH10Bac) that contain a bacmid with a transposition site. Through site-specific transposition, the gene of interest is moved from the donor plasmid into the bacmid, creating a recombinant bacmid. White/blue selection is used to identify positive colonies. The recombinant bacmid DNA is then isolated [17].
- Virus Generation and Titration (Steps 5-7): The isolated bacmid DNA is transfected into insect cells (e.g., Sf9) to produce the initial (P1) recombinant baculovirus stock. This P1 stock is amplified by infecting a fresh culture of log-phase insect cells at a low multiplicity of infection (MOI of 0.05-0.1) to generate a higher-titer P2 stock. The viral titer is determined via a plaque assay to ensure reproducible infection conditions. A titer of ≥1 x 10^8^ PFU/mL is recommended for expression studies [22].
- Protein Expression (Steps 8-9): For large-scale protein production, healthy log-phase insect cells are infected with the amplified, titered virus at an optimal MOI (typically 5-10). Cells are harvested 48-72 hours post-infection, at the peak of recombinant protein production, after which cellular processes may break down [22]. The recombinant protein is then purified from the cell culture supernatant or lysate using appropriate chromatographic methods.
The Scientist's Toolkit: Essential Research Reagents
A successful insect cell culture and protein expression workflow relies on a set of core reagents and cell lines.
Table 3: Essential Research Reagents for Insect Cell-Based Protein Production
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Insect Cell Lines | Host for baculovirus infection and recombinant protein production. | Sf9/Sf21: General purpose, virus amplification. High Five: High yield for secreted proteins [17]. |
| Serum-Free Media (SFM) | Supports high-density growth in suspension; defined formulation. | Gibco Sf-900 II/III SFM for Sf cells; Express Five SFM for High Five cells [22]. |
| Baculovirus Expression System | Vector for delivering gene of interest to insect cells. | Bac-to-Bac (most common), BaculoDirect, Bac-N-Blue [22]. |
| Dissociation Reagent | Detaches adherent cells for subculturing and passage. | Trypsin or animal-origin-free TrypLE [77]. |
| Baculovirus Titer Kit | Quantifies infectious virus particles for reproducible experiments. | Plaque assay kits or faster qPCR-based titer kits. |
| Bioreactor / Suspension Culture System | Enables scalable production from small to large volumes. | Spinner flasks, shake flasks, or stirred-tank bioreactors. |
| Shear-Force Protectant | Protects cells from damage in suspension culture. | Pluronic F-68, often included in serum-free media [22]. |
The insect cell expression system has proven to be a robust, scalable, and economically viable platform for producing some of the most critical modern vaccines and therapeutics. The case studies of NVX-CoV2373 and Cervarix demonstrate its capacity to meet stringent regulatory standards and address global health challenges. The provided protocols for subculturing adherent cells and generating recombinant proteins offer a foundational guide for researchers to leverage this powerful technology. As genetic engineering continues to enhance the glycosylation pathways and resilience of insect cell lines, the scope and efficiency of this platform are poised for further expansion, solidifying its role in the future of biomanufacturing [14] [13] [17].
Conclusion
Mastering the subculture of adherent insect cells is fundamental for leveraging their full potential in biopharmaceutical production and advanced research. This guide has synthesized key principles, from foundational biology and meticulous protocols to proactive troubleshooting and system comparisons. The future of this field points toward further optimization through genetic engineering of cell lines, refined animal-free media formulations, and the development of more scalable hybrid systems like microcarriers. Adopting these robust and reproducible practices will directly contribute to advancements in producing complex biologics, vaccines, and novel therapeutics, solidifying the role of insect cell culture as a pillar in modern biomedical science.
References
- 1. Adherent Cell Culture Protocol [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-adherent-cells.html]
- 2. Advances in cell culture: anchorage dependence - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4275909/]
- 3. Insect Cell Culture - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/insect-cell-culture]
- 4. Cell culture and maintenance protocol [https://www.abcam.com/en-us/technical-resources/protocols/cell-culture-and-maintenance-protocol]
- 5. Methods for Maintaining Insect Cell Cultures - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC355909/]
- 6. Adherent Cell Culture Bioreactors | Esco Healthcare [https://escovaccixcell.com/Tide_Technology]
- 7. Sf21 [https://www.culturecollections.org.uk/nop/product/sf21]
- 8. The Bioengineering of Insect Cell Lines for Biotherapeutics ... [https://www.mdpi.com/2076-393X/13/6/556]
- 9. Sf9 Cell Line - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/sf9-cell-line]
- 10. A beginners guide to Sf9 and Sf21 insect cell line culture and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12144304/]
- 11. Sf9 Cells [https://www.cytion.com/us/Sf9-Cells/604329]
- 12. High Five cells [https://en.wikipedia.org/wiki/High_Five_cells]
- 13. Insect cell expression system: advances in applications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12502203/]
- 14. The Bioengineering of Insect Cell Lines for Biotherapeutics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12197355/]
- 15. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7159335/]
- 16. Baculovirus Display of Peptides and Proteins for Medical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9962271/]
- 17. A beginners guide to Sf9 and Sf21 insect cell line culture ... [https://www.nature.com/articles/s41598-025-99812-0]
- 18. Baculovirus-free insect cell expression system for high ... [https://www.nature.com/articles/s41598-020-78425-9]
- 19. Insect cell expression system: advances in applications ... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-025-00562-4]
- 20. Baculovirus Display of Peptides and Proteins for Medical ... [https://www.mdpi.com/1999-4915/15/2/411]
- 21. Improving the production of baculovirus expression vector by ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0320182]
- 22. Insect Cell–Based Protein Expression [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/protein-expression-handbook/pex-handbook-insect-cell-based-protein-expression.html]
- 23. What are the advantages of the insect protein expression ... [https://www.aatbio.com/resources/faq-frequently-asked-questions/what-are-the-advantages-of-the-insect-protein-expression-system]
- 24. Cost Efficient R&D Scale Up for Insect Cell Recombinant ... [https://www.corning.com/worldwide/en/products/life-sciences/resources/stories/at-the-bench/cost-efficient-rd-scale-up-for-insect-cell-recombinant-protein-production.html]
- 25. Exploring the Unique Characteristics of Insect Cell Lines [https://cellculturecompany.com/exploring-the-unique-characteristics-of-insect-cell-lines/]
- 26. The Coming Age of Insect Cells for Manufacturing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6420222/]
- 27. Evaluation of an inducible knockout system in insect cells ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10374150/]
- 28. Suspension Cell Culture Protocol | Thermo Fisher Scientific [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-suspension-cells.html]
- 29. High shear resistance of insect cells: the basis for ... [https://www.nature.com/articles/s41598-021-88813-4]
- 30. Optimizing HEK293 cell cultures for gene therapy ... [https://infors-ht.com/en/blog/optimizing-hek293-cell-cultures-for-gene-therapy-applications-the-role-of-incubator-flexibility]
- 31. Advances in Insect Cell-Expression Systems [https://www.biopharminternational.com/view/advances-in-insect-cell-expression-systems]
- 32. Dissociation Protocols [https://www.cytion.com/us/Knowledge-Hub/Cell-Culture-Techniques/Dissociation-Protocols/]
- 33. Adherent & Suspension Cell Culture in Bioprocessing [https://escovaccixcell.com/news/adherent-suspension-cell-culture-in-bioprocessing]
- 34. Insect Cell Culture Media Analysis 2025 and Forecasts 2033 [https://www.archivemarketresearch.com/reports/insect-cell-culture-media-283127]
- 35. How Insect Cell Culture Medium Works — In One Simple ... [https://www.linkedin.com/pulse/how-insect-cell-culture-medium-works-one-simple-flow-2025-pzwwe/]
- 36. Different methods of detaching adherent cells and their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8983651/]
- 37. Enzymatic and Non-Enzymatic Cell Dissociation Protocols [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/cell-dissociation-protocol.html]
- 38. Cell Dissociation Methods for Disaggregation of Tissue [https://www.akadeum.com/blog/cell-dissociation/?srsltid=AfmBOooV0bjxGgqR8TqmBC1KwfKFM2P1N1aEjUCSbbvFNNkumqJeJa0K]
- 39. Supporting the First US FDA Approval of Serum-Free Stem ... [https://www.sgs.com/en/news/2025/10/supporting-the-first-us-fda-approval-of-serum-free-stem-cell-culture-media-in-china]
- 40. The Development and Impact of Serum-Free Media [https://pubmed.ncbi.nlm.nih.gov/41074929/]
- 41. Improving the human relevance of cell culture using animal ... [https://nc3rs.org.uk/3rs-resources/improving-human-relevance-cell-culture-using-animal-free-culture-media]
- 42. Comparison of culture media supplements identifies serum ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-025-04561-6]
- 43. Animal Component Free Media & Reagents Frequently ... [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/cell-culture-and-cell-culture-analysis/cell-culture-media-preparation/animal-component-free-frequently-asked-questions?srsltid=AfmBOoppIg9fzAgdhd0Eq85MpsdAL4ujiNn3SBGPbgKR1E1JJGiBuEI4]
- 44. Preparation of a universally usable, animal product free, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10912720/]
- 45. ATCC Animal Cell Culture Guide [https://www.atcc.org/resources/culture-guides/animal-cell-culture-guide]
- 46. Insect Cell Culture Support—Getting Started [https://www.thermofisher.com/us/en/home/technical-resources/technical-reference-library/cell-culture-support-center/insect-cell-culture-support/insect-cell-culture-support-getting-started.html]
- 47. Suspension Cell Culture Protocol [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-suspension-cells.reg.sg.html]
- 48. A Beginner's Guide to Cell Culture: Practical Advice for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10000895/]
- 49. The Simple Ways to Prevent Cell Culture Contamination [https://www.fishersci.com/us/en/science-social-hub/buying-guides/the-simple-ways-prevent-cell-culture-contamination.html]
- 50. Cell Culture Contamination: How to Prevent Fungal, Yeast, ... [https://www.corning.com/worldwide/en/products/life-sciences/resources/stories/at-the-bench/how-to-control-bacterial-contamination-and-other-unwelcome-micro.html]
- 51. Cell Culture Contamination | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/biological-contamination.html]
- 52. Morphology of Sf21 and Sf9 Cells [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-expression-information/morphology-sf21-sf9-cells.html]
- 53. I have an insect cell line, Sf21 and I need to transition to serum free medium. I have tried to do this before but the growth rate decreased from 24 to 50 hours. I have new cells so what should I do. [https://cellculturedish.com/questions/i-have-an-insect-cell-line-sf21-and-i-need-to-transition-to-serum-free-medium-i-have-tried-to-do-this-before-but-the-growth-rate-decreased-from-24-to-50-hours-i-have-new-cells-so-what-should-i-do/]
- 54. Optimization of the growth conditions of Sf21 insect cells for high-density perfusion culture in stirred-tank bioreactors - PubMed [https://pubmed.ncbi.nlm.nih.gov/7764890/]
- 55. Development of an Optimized Process for Functional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10674791/]
- 56. A new single-step protocol for rapid baculovirus-driven protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5689143/]
- 57. Relevant principal factors affecting the reproducibility of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5486689/]
- 58. Improving small-scale cultivation of Spodoptera frugiperda ... [https://www.nature.com/articles/s41598-024-84093-w]
- 59. The untapped potential of cell culture in disentangling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11149091/]
- 60. How to Reduce Cell Clumping in Single Cell Suspensions [https://www.stemcell.com/how-to-reduce-cell-clumping-in-single-cell-suspensions-with-dnase.html]
- 61. How to Reduce Cell Clumping in Cell Cultures [https://www.akadeum.com/blog/cell-clumping/?srsltid=AfmBOooy8YwErhjmGGWs-ZZ3Wwue4QgXAbL0Zom_KoS_xjpnKGmp2zrA]
- 62. Best Practices for Preparing a Single Cell Suspension from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6375754/]
- 63. How To Prepare a Good Single Cell Suspension [https://www.appliedcytometry.com/articles/good-single-cell-suspension/]
- 64. Insect Cell Lines Market Size, Demand & Trends 2025-2035 [https://www.futuremarketinsights.com/reports/insect-cell-lines-market]
- 65. Adherent Cell Culture Protocol [https://www.thermofisher.com/tw/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-adherent-cells.html]
- 66. Monitoring of adherent live cells morphology using the ... [https://pubmed.ncbi.nlm.nih.gov/27477880/]
- 67. Insect Cell Medium Innovations Shaping Market Growth ... [https://www.datainsightsmarket.com/reports/insect-cell-medium-948617]
- 68. Seed Train Optimization in Microcarrier-Based Cell Culture ... [https://www.mdpi.com/2306-5354/11/3/268]
- 69. What Are the Key Characteristics of Mammalian vs Insect ... [https://synapse.patsnap.com/article/what-are-the-key-characteristics-of-mammalian-vs-insect-cell-lines]
- 70. Production of Virus in Insect Versus Mammalian Cells [https://blog.addgene.org/production-of-virus-in-insect-vs-mammalian-cells]
- 71. Guidelines to Maintain Cultured Cells [https://www.thermofisher.com/jp/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/maintaining-cultured-cells.html]
- 72. Establishment & characterization of a non-adherent insect cell line for cultivated meat | Scientific Reports [https://www.nature.com/articles/s41598-025-86921-z]
- 73. Microcarriers as a culturing system of insect cells and insect viruses - PubMed [https://pubmed.ncbi.nlm.nih.gov/2438174/]
- 74. Microcarrier Technology: Advancing Adherent Cell Therapies [https://ritaibioreactor.com/adherent-cell/]
- 75. Seed Train Optimization in Microcarrier-Based Cell Culture ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10968011/]
- 76. BioNOC II® Cell Culture Macrocarriers | Esco Healthcare [https://escovaccixcell.com/tide_technology/cell_attachment/BioNOC_II]
- 77. Culture Adherent | Thermo Fisher Scientific - CN Cell Protocol [https://www.thermofisher.com/cn/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/subculturing-adherent-cells.html]