Balancing Act: How Freezing Rates Govern Cell Dehydration and Ice Formation in Biopreservation
This article provides a comprehensive analysis of the critical role freezing rates play in cryopreservation, directly impacting cell dehydration and ice formation—two primary drivers of cell death.
Balancing Act: How Freezing Rates Govern Cell Dehydration and Ice Formation in Biopreservation
Abstract
This article provides a comprehensive analysis of the critical role freezing rates play in cryopreservation, directly impacting cell dehydration and ice formation—two primary drivers of cell death. Tailored for researchers, scientists, and drug development professionals, we explore the fundamental biophysical principles, detail methodological approaches for process control, present strategies for troubleshooting and optimization, and review validation techniques for comparative analysis. By synthesizing foundational knowledge with the latest advancements, including controlled ice nucleation, this resource aims to equip professionals with the insights needed to develop robust, high-yield preservation protocols for cell therapies and biopharmaceuticals.
The Ice-Water Dilemma: Foundational Principles of Cellular Cryoinjury
Cryopreservation stands as an indispensable technology in biomedical fields, particularly for the emerging cell and gene therapy industry, where it enables long-term storage and off-the-shelf availability of living cellular products. The fundamental challenge lies in navigating the delicate balance between two competing injury mechanisms that occur when cells are subjected to subzero temperatures. As Mazur's "two-factor hypothesis" established, cellular survival during freezing and thawing depends critically on avoiding both intracellular ice formation and excessive dehydration, with the cooling rate serving as the primary determinant of which mechanism predominates [1] [2]. This whitepaper provides an in-depth technical examination of these competing injury mechanisms, framed within contemporary research on how freezing rates govern the complex interplay between cellular dehydration and ice formation.
The high stakes of optimizing cryopreservation protocols are evident in the clinical realm, where cryopreservation failure can result in substantial loss of post-thaw cell viability and function, potentially compromising therapeutic efficacy [3]. For cell therapy products like CAR-T cells, the cryopreservation process significantly influences the safety, reliability, and effectiveness of these revolutionary treatments [4]. A comprehensive understanding of cryoinjury mechanisms thus represents not merely an academic pursuit but a critical requirement for advancing clinical applications of cellular therapies.
Theoretical Framework: The Two-Factor Hypothesis of Cryoinjury
The Physical-Chemical Basis of Freezing Injury
When an aqueous cell suspension is cooled below its freezing point, water molecules begin to crystallize into ice in the extracellular space first. This initial ice formation creates a chemical potential gradient that drives intracellular water out of the cell through an osmotically driven process. The remaining extracellular solution becomes increasingly concentrated with solutes, leading to elevated osmotic pressure that progressively dehydrates the cell [4] [5]. The competing risks during this process are straightforward in concept but complex in practice: too little dehydration results in intracellular ice formation, while too much dehydration causes solution-effects injury.
The relationship between cooling rate and cell survival follows a characteristic inverted U-shape curve, where an optimal cooling rate exists that minimizes both forms of cryoinjury. At slow cooling rates (typically <1°C/min for many mammalian cells), cells experience extensive dehydration, avoiding intracellular ice formation but potentially suffering from solute effects injury and excessive volumetric reduction. At rapid cooling rates (typically >10°C/min), insufficient time for water export leads to supercooled cytoplasm that eventually results in lethal intracellular ice formation [1] [2]. The precise optimal rate varies significantly between cell types based on their membrane permeability characteristics and surface-to-volume ratios.
Quantitative Modeling of Cellular Responses
Theoretical models of cellular responses to freezing have become increasingly sophisticated, incorporating parameters such as water permeability, its temperature dependence (activation energy), and the osmotically inactive volume fraction of cells. Simulations can predict changes in intracellular water volume as a function of temperature during cooling, providing valuable indicators for predicting cryoinjury. Intracellular supercooling has been established as a reliable indicator of intracellular ice formation potential, while intracellular osmolality serves as an indicator of solution effects injury [2].
Table 1: Key Parameters in Cryoinjury Simulation Models
| Parameter | Symbol | Role in Cryoinjury Prediction | Experimental Determination |
|---|---|---|---|
| Water permeability | Lp | Determines rate of cellular dehydration during cooling | Cryomicroscopy with volume measurements |
| Activation energy | Ea | Temperature dependence of water permeability | Arrhenius plot of Lp vs. temperature |
| Osmotically inactive volume | Vb | Fraction of cell volume not participating in osmosis | Boyle van't Hoff plot |
| Intracellular supercooling | ΔT | Indicator of intracellular ice formation risk | Calculated from intracellular freezing point |
| Intracellular osmolality | Os | Indicator of solute effects injury | Calculated from solute concentrations |
These parameters enable researchers to simulate cellular responses under various freezing conditions and design optimized protocols before empirical testing. For instance, simulations of TF-1 cells during graded freezing experiments have demonstrated that high intracellular osmolality (>4 Osm) correlated with solution effects injury in slow-cooled samples, while intracellular supercooling exceeding 10°C predicted intracellular ice formation upon plunging into liquid nitrogen [2].
Mechanism I: Intracellular Ice Formation (IIF)
Mechanisms and Consequences of IIF
Intracellular ice formation represents a predominantly lethal event during cryopreservation, occurring when the cooling rate exceeds a cell's capacity to dehydrate in response to extracellular freezing. As the extracellular solution freezes, intracellular water becomes supercooled - remaining liquid despite being below its thermodynamic freezing point. When this supercooling reaches a critical threshold (typically between -5°C and -15°C below the intracellular freezing point), the homogeneous or heterogeneous nucleation of ice crystals within the cytoplasm occurs, often with catastrophic results [2].
The destructive capacity of intracellular ice stems from both mechanical and biochemical damage mechanisms. Ice crystals can physically disrupt organelles, the cytoskeleton, and the plasma membrane through direct mechanical action [5]. This is visually evident under microscopy as a darkening of cellular appearance following IIF [4]. The formation of intracellular ice also creates concentration gradients within the cell that can lead to membrane rupture during thawing. Research using cryomicroscopy has consistently demonstrated that cells exhibiting intracellular ice formation rarely maintain membrane integrity or viability post-thaw.
Experimental Detection and Analysis
Detection of intracellular ice formation employs both direct and indirect methodologies. Cryomicroscopy allows direct visualization of ice formation in real-time during freezing, with IIF manifesting as a sudden darkening or change in light scattering properties within the cell [4]. Alternatively, graded freezing experiments provide an indirect assessment where cells are cooled slowly to various subzero temperatures before being plunged into liquid nitrogen. Significantly reduced viability in plunged samples compared to directly thawed ones indicates intracellular ice formation occurring during the rapid cooling phase [2].
Recent investigations have employed fluorescence microscopy in combination with polarized light to simultaneously monitor ice formation and cell volume changes, providing unprecedented insight into the dynamics of IIF. These approaches have revealed that the temperature of extracellular ice nucleation significantly influences IIF incidence, with controlled nucleation at higher subzero temperatures (e.g., -6°C) reducing supercooling and consequently decreasing IIF probability [4].
Table 2: Experimental Approaches for Studying Intracellular Ice Formation
| Methodology | Key Measurements | Advantages | Limitations |
|---|---|---|---|
| Cryomicroscopy | Visual observation of ice formation | Direct, real-time observation | Limited to small samples |
| Graded freezing | Viability comparison between direct-thaw and plunged samples | Quantifies temperature zones of IIF risk | Post-thaw assessment only |
| Fluorescence microscopy with membrane integrity stains | Correlation of IIF with membrane damage | Simultaneous assessment of multiple parameters | Complex setup and analysis |
| Differential scanning calorimetry (DSC) | Measurement of heat release during IIF | Quantitative, population-level data | Does not assess individual cells |
Mechanism II: Excessive Dehydration and Solute Effects
The Dehydration Process and Its Consequences
In contrast to the rapid damage of intracellular ice formation, injury from excessive dehydration occurs gradually during slow cooling as cells lose water to the increasingly concentrated extracellular solution. As extracellular ice forms, solutes are excluded from the growing ice crystals, creating a hypertonic extracellular environment that drives osmotic water efflux from cells. This process continues until temperatures drop sufficiently to vitrify the highly concentrated intracellular solution, typically below -40°C [4] [5].
The injurious effects of excessive dehydration manifest through multiple mechanisms. Volumetric reduction can reach dramatic proportions, with cells losing up to 90% of their water content, potentially leading to membrane collapse and irreversible structural damage [5]. The concentrated intracellular solutes can denature proteins, disrupt metabolic functions, and alter lipid bilayer properties. Additionally, prolonged exposure to highly concentrated solutions can cause membrane damage through a process known as "solution effects" injury, where the altered chemical environment directly compromises membrane integrity [2].
Experimental Assessment of Dehydration Injury
Investigating dehydration injury requires methodologies distinct from those used for intracellular ice formation. Cell volume tracking during freezing, often via cryomicroscopy with image analysis, provides direct measurement of dehydration kinetics [4]. Alternatively, manipulating cooling rates and observing differential survival can indicate dehydration injury, with poor recovery at very slow cooling rates suggesting solute effects as the dominant injury mechanism [1].
Recent research has employed controlled ice nucleation to standardize the dehydration process across samples. Studies on Jurkat cells (a model for T-cells) have demonstrated that initiating extracellular ice formation at a higher temperature (-6°C versus -10°C) promotes more gradual dehydration, reduces intracellular ice formation, and improves post-thaw recovery, particularly in lower concentrations of cryoprotectants [4]. This approach highlights the critical relationship between ice nucleation temperature and dehydration kinetics.
The Critical Role of Freezing Rate
Cooling Rate as the Balancing Factor
The freezing rate serves as the primary determinant of which cryoinjury mechanism predominates, creating a fundamental trade-off between dehydration and intracellular ice formation. At slow cooling rates (approximately 1°C/min for many mammalian cells), sufficient time exists for cellular dehydration to follow equilibrium conditions, minimizing intracellular ice formation but potentially resulting in excessive dehydration and solute damage. At rapid cooling rates (>10-100°C/min, depending on cell type), insufficient time for water export leads to supercooled cytoplasm that eventually nucleates intracellular ice [4] [5] [2].
The concept of an "optimal cooling rate" emerges from this balance, representing the cooling velocity that minimizes combined injury from both mechanisms. This optimal rate varies substantially between cell types based on their biophysical properties, particularly membrane water permeability and surface-to-volume ratio. Cells with higher water permeability can tolerate faster cooling rates because they dehydrate more rapidly in response to extracellular freezing.
Advanced Cooling Rate Concepts
Recent research has revealed nuances beyond the basic two-factor hypothesis, including the potential benefits of non-linear cooling profiles. Some protocols employ an initial slow cooling rate (e.g., 1°C/min) to promote dehydration, followed by faster cooling (e.g., 10°C/min) once significant dehydration has occurred, thereby reducing processing time while maintaining viability [4].
The influence of ice nucleation temperature further complicates the simple cooling rate paradigm. Studies demonstrate that controlling the initiation of extracellular ice formation at specific temperatures significantly impacts subsequent dehydration kinetics and intracellular ice formation probability. For Jurkat cells, controlled nucleation at -6°C rather than -10°C resulted in enhanced cellular dehydration with less incidence of intracellular ice formation, particularly in formulations with reduced cryoprotectant concentrations [4].
Experimental Approaches and Methodologies
Core Techniques in Cryoinjury Research
Investigating cryoinjury mechanisms requires specialized methodologies capable of monitoring cellular responses during the dynamic freezing process. The following experimental approaches represent current standards in the field:
Cryomicroscopy with fluorescence capabilities enables real-time observation of ice formation, cell volume changes, and membrane integrity during controlled freezing. This technique typically involves a temperature-controlled stage coupled with brightfield and fluorescence microscopy, allowing correlation of physical events (ice formation) with biological outcomes (membrane damage) [4]. Samples are prepared as thin films between coverslips to ensure uniform temperature distribution, with cells often stained with viability indicators such as acridine orange and propidium iodide.
Graded freezing experiments systematically investigate the temperature dependence of both injury mechanisms. This approach involves cooling cell suspensions slowly (e.g., 1°C/min) to various target subzero temperatures, with aliquots either thawed directly (assessing dehydration injury) or plunged into liquid nitrogen before thawing (assessing intracellular ice formation injury) [2]. The differential survival between these conditions reveals the temperature zones where each injury mechanism predominates.
Controlled ice nucleation techniques standardize the initial freezing event to reduce sample variability. Methods include manual ice seeding, shock cooling, chemical nucleates, and pressure shift technology [4]. The pressurization/depressurization method (e.g., Control Lyo) represents a recent advancement that induces ice nucleation at defined temperatures without liquid nitrogen, improving process consistency.
Post-Thaw Assessment Methods
Accurate evaluation of cryoinjury requires multidimensional assessment of cell status after thawing. Membrane integrity assays using dye exclusion (e.g., trypan blue, propidium iodide) represent the most common viability measure but provide limited information as they detect only severe membrane damage [5]. Functional assessments including metabolic activity assays, clonal growth potential, and cell-specific functions (e.g., immunosuppressive capacity for T-cells) offer more physiologically relevant recovery indicators [3]. Advanced omics technologies including proteomics and metabolomics have revealed molecular-level damage undetectable by conventional methods, identifying oxidative stress responses and energy pathway disruptions in cryoinjured cells [6].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents and Experimental Materials for Cryoinjury Studies
| Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Cell Models | Jurkat cells (T-cell model) [4], TF-1 cells [2], CHO cells [1], MSCs [3] | Provide biologically relevant systems for cryoinjury studies | Primary cells often more sensitive than immortalized lines |
| Cryoprotectants | Dimethyl sulfoxide (DMSO) [4], Trehalose [1], Glycerol [5] | Protect cells from freezing injury via multiple mechanisms | DMSO concentration typically 5-10%; concerns about toxicity |
| Assessment Reagents | Acridine Orange [4], Propidium Iodide [4], Trypan Blue [5] | Evaluate membrane integrity and viability | Fluorescent stains enable real-time monitoring during freezing |
| Specialized Equipment | Controlled Rate Freezers [4] [7], Cryomicroscopy Systems [4] [2], DSC Instruments [8] | Enable precise temperature control and real-time observation | CRFs allow documentation for cGMP manufacturing |
| Ice Nucleation Agents | Chemical nucleates, Pressure shift technology (Control Lyo) [4] | Standardize initial ice formation at defined temperatures | Reduces sample variability in freezing studies |
Implications for Cell Therapy Development
The principles of cryoinjury mechanisms directly impact the burgeoning field of cell therapy, where post-thaw viability and functionality represent critical quality attributes. Current industry surveys indicate that 87% of cell therapy developers employ controlled-rate freezing, with 60% using default cooling profiles despite recognition that optimized profiles may be necessary for sensitive cell types including iPSCs, CAR-T cells, and differentiated derivatives [7].
The toxicity concerns surrounding DMSO, the most common cryoprotectant, have driven research toward reduced concentration formulations (2.5-5% versus conventional 10%). Success with these formulations often requires precise manipulation of freezing parameters, particularly controlled ice nucleation and optimized cooling rates, to compensate for reduced cryoprotection [4] [9]. This approach highlights the interplay between cryoprotectant concentration and freezing protocol optimization in mitigating cryoinjury.
For allogeneic (off-the-shelf) cell therapies, where products are cryopreserved at scale and shipped to clinical sites, understanding and controlling cryoinjury mechanisms becomes essential for product consistency and patient safety. In these applications, the complete cryopreservation process - including freezing parameters, storage conditions, and thawing methods - must be rigorously controlled to ensure predictable post-thaw potency [9] [7].
The intricate balance between intracellular ice formation and excessive dehydration continues to define fundamental challenges in cryopreservation science. While Mazur's two-factor hypothesis provides a robust theoretical framework, contemporary research has revealed additional layers of complexity, including the significant roles of ice nucleation temperature, non-linear cooling profiles, and cell-type-specific biophysical properties. The optimization of cryopreservation protocols for emerging cell therapies demands sophisticated approaches that account for these variables through controlled experimentation and computational modeling.
Future directions in cryoinjury research will likely focus on personalized freezing protocols tailored to specific cell types, the development of DMSO-free cryopreservation solutions that maintain efficacy without toxicity concerns, and the implementation of advanced process analytics to ensure consistent product quality. As cryopreservation remains a critical enabling technology for cellular therapies and regenerative medicine, continued elucidation of cryoinjury mechanisms will directly contribute to the successful clinical translation of these revolutionary treatments.
The kinetics of water transport during freezing are a critical determinant of cell survival and post-thaw quality in fields ranging from clinical cryopreservation to food science. The rate at which cells are cooled governs the physical movement of water across cell membranes, which in turn dictates whether water remains intracellularly to form damaging ice crystals or exits the cell, leading to protective dehydration. This whitepaper synthesizes current research on the biophysical principles underlying water transport kinetics, providing a technical guide for researchers and scientists working in drug development and cellular biology. Understanding these mechanisms is essential for developing optimized cryopreservation protocols that maintain cell viability and function.
Theoretical Foundations: Water Transport and Freezing Kinetics
Fundamental Biophysical Principles
When a cell suspension is cooled below its freezing point, extracellular water begins to crystallize first. This ice formation excludes solutes, increasing the osmotic pressure of the remaining extracellular fluid. Consequently, an osmotic gradient is established across the cell membrane, driving intracellular water outward in an attempt to restore equilibrium. The extent and kinetics of this water efflux are governed by the cooling rate, membrane permeability to water, and the surface area-to-volume ratio of the cell [10] [8].
The movement of water across the cell membrane during freezing is an osmotically driven process that can be mathematically described by membrane transport models. These models account for the temperature-dependent permeability of the cell membrane, typically characterized by the membrane hydraulic conductivity (Lpg) and its activation energy (ELp) [11]. At slow cooling rates (e.g., -1°C/min), water has sufficient time to exit the cell, leading to significant cell dehydration and shrinkage. This dehydration concentrates intracellular solutes, potentially reaching toxic levels and causing so-called "solution effects" damage. Conversely, at rapid cooling rates (e.g., -10°C/min or faster), water does not have sufficient time to exit the cell before intracellular temperatures reach a point where the remaining supercooled water freezes, resulting in lethal intracellular ice formation [12].
The Role of Ice Crystal Dynamics
Ice crystal formation and growth are intimately connected to water transport kinetics. During slow freezing, the progressive migration of water from intracellular to extracellular spaces results in the formation of large, dendritic ice crystals in the extracellular matrix. These crystals can mechanically disrupt tissue architecture and cell membranes [13]. Experimental studies on strawberry tissue have demonstrated that slow freezing rates (1.57 ± 0.01 cm/h) cause severe cellular structure damage compared with faster freezing rates (10.43 ± 0.35 cm/h), with relative conductivity increasing by approximately 40% compared with fresh samples, indicating membrane disruption [14].
Rapid cooling promotes the formation of numerous small ice crystals that are more uniformly distributed both inside and outside cells. This finer ice crystal structure is associated with better preservation of cellular ultrastructure and tissue integrity [14] [13]. The relationship between cooling rate and ice crystal size has been quantitatively demonstrated through fractal dimension analysis, with frozen treatment groups showing approximately 9% increases in fractal dimension compared with fresh samples [14].
Table 1: Impact of Cooling Rate on Cellular Water States and Ice Crystals
| Cooling Rate | Bound Water Conversion | Ice Crystal Morphology | Cellular Structural Damage |
|---|---|---|---|
| Slow (~1.6°C/min) | Significant conversion to free water (≈4% increase) [14] | Large, uneven crystals with dendritic growth [13] | Severe membrane damage; ≈40% increase in relative conductivity [14] |
| Rapid (~10°C/min) | Minimal water state conversion [14] | Small, uniform crystals distributed evenly [14] [13] | Preservation of original cell structure; minimal membrane disruption [14] |
Quantitative Data on Cooling Rate Effects
Systematic Studies in Biological Systems
The interaction between cooling rate and cell viability has been systematically investigated across different cell types. Research on human peripheral blood T cells has revealed a critical interaction between cooling and warming rates. When cells were cooled slowly (-1°C/min), the warming rate had minimal impact on viable cell recovery across a wide range of thawing rates (1.6°C/min to 113°C/min). However, with rapid cooling (-10°C/min), slow warming (1.6°C/min and 6.2°C/min) resulted in significant cell death, while rapid warming (45°C/min and 113°C/min) maintained viability. This phenomenon was correlated with ice recrystallization events observed during slow thawing following rapid freezing [12].
Cryomicroscopy studies of MCF7 breast cancer cells have provided quantitative parameters for water transport kinetics, with average biophysical parameters across cooling rates of 5, 10, and 20°C/min determined to be Lpg = 0.10 μm/min-atm and ELp = 15.5 kcal/mol [11]. These parameters allow for predictive modeling of water transport during freezing and have been validated through comparison with direct cryomicroscopy observations [11].
Recent research on Jurkat T cells has further elucidated the impact of different freezing phases, demonstrating that not just the cooling rate but specifically the rate of ice crystal formation emerged as a crucial factor, with viability ranging from 0.3% to 53.2% depending on this parameter [15].
Table 2: Cell Type-Specific Responses to Cooling Rate
| Cell/Tissue Type | Optimal Cooling Rate | Key Findings | Viability/Quality Range |
|---|---|---|---|
| Jurkat T Cells | Variable by phase | Ice crystal formation rate most critical factor [15] | 0.3%-53.2% (varies with formulation) [15] |
| Human PB T Cells | -1°C/min | Viability maintained across warming rates; rapid cooling requires rapid warming [12] | High viability maintained with proper cooling/warming combination [12] |
| MCF7 Cells | Modeled for multiple rates | Water transport parameters established: Lpg=0.10 μm/min-atm, ELp=15.5 kcal/mol [11] | Parameters validated with cryomicroscopy [11] |
| Strawberry Tissue | 10.43 ± 0.35 cm/h | Minimal cellular damage; limited water state conversion [14] | Best preservation of cellular structure [14] |
Experimental Methodologies and Protocols
Differential Scanning Calorimetry (DSC) for Water Transport Measurement
DSC provides a quantitative method for measuring water transport during freezing in cell suspensions. The technique exploits the heat release differences between samples containing osmotically active cells and identical samples with osmotically inactive or lysed cells [11].
Protocol Details:
- Sample Preparation: Prepare cell suspensions at appropriate cytocrit (cell concentration) in culture media. Control samples should include osmotically inactive cells (achieved through lysing or specific treatment).
- Instrument Calibration: Calibrate the DSC instrument using standard references according to manufacturer specifications. Ensure temperature accuracy, particularly in the sub-zero range.
- Freezing Protocol: Load samples into DSC pans and initiate controlled cooling programs. Typical cooling rates for water transport studies range from 5°C/min to 20°C/min [11].
- Data Analysis: Calculate the difference in heat release (Δqdsc) between active and inactive cell samples. This value is proportional to the cytocrit and the supercooled water volume in the sample. Plot normalized fractional integrated heat release difference (Δq(T)dsc/Δqdsc) as a function of temperature to correlate with the amount of supercooled cellular water that has exosmosed from the cell at different subzero temperatures [11].
Limitations and Considerations:
- The technique requires a priori knowledge of geometric parameters including surface area, initial volume, and osmotically inactive cell volume [11].
- DSC alone cannot distinguish whether heat release from supercooled cellular water is due to dehydration or intracellular ice formation, necessitating complementary techniques like cryomicroscopy for verification [11].
Cryomicroscopy for Direct Visualization
Cryomicroscopy allows direct observation of cellular responses during freezing, providing visual confirmation of dehydration kinetics and intracellular ice formation.
Protocol Details:
- Sample Preparation: Place cell suspension on a temperature-controlled microscope stage equipped with a specialized freezing chamber.
- Temperature Programming: Implement controlled cooling protocols matching those used in DSC studies. Typical rates range from 5°C/min to 20°C/min [11].
- Image Acquisition: Capture time-lapse images throughout the freezing process, focusing on cell volume changes and ice formation events.
- Volume Analysis: Measure cell volume changes during freezing by tracing cell boundaries in captured images. Compare with theoretical models of water transport.
- Intracellular Ice Detection: Identify intracellular ice formation by the sudden darkening or appearance of ice within cell boundaries [11].
Validation Studies: Cryomicroscopy has been used to validate DSC water transport data, with studies showing strong statistical correlation between the two techniques at cooling rates of 5°C/min [11]. This correlation confirms that dehydration is the predominant biophysical response at conventional cooling rates, with intracellular ice formation becoming significant only at higher cooling rates (e.g., 20°C/min) [11].
Low-Field Nuclear Magnetic Resonance (LF-NMR) for Water State Analysis
LF-NMR spectroscopy provides a non-destructive method for monitoring water states and distribution during freezing processes by measuring T2 relaxation times [14] [13].
Application in Tissue Studies: Research on strawberries has employed LF-NMR to demonstrate that freezing changes the relaxation time of different water states, with slow freezing rates (1.57 ± 0.01 cm/h) causing bound water to be more easily converted to free water [14]. This conversion promotes significant water loss, with the proportion of free water in frozen samples increasing by approximately 4% compared with fresh samples [14].
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Materials for Water Transport Studies
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| Permeating Cryoprotectants | Reduce ice crystal formation; enable vitrification; depress freezing point [10] | DMSO (10% common concentration), Glycerol, Ethylene Glycol, Propylene Glycol [10] |
| Non-Permeating Agents | Provide extracellular cryoprotection; enable reduced permeating agent concentrations [10] | Sucrose, Trehalose, Raffinose, PVP, PEG [10] |
| Cell Viability Assays | Assess post-thaw cell survival and function [12] | Trypan Blue exclusion, Flow cytometry with live/dead staining, Functional assays [12] |
| Engineered Tissue Constructs | Model cell-fluid-matrix interactions during freezing [8] | Collagen matrices with embedded cells (e.g., MCF7 breast cancer cells) [8] |
| Temperature-Controlled Stages | Precisely control cooling/warming rates for microscopy [11] | Linkam MDS 600, Custom-built systems with temperature reservoirs [11] [8] |
Visualizing Water Transport Kinetics
The kinetics of water transport during freezing represent a critical balance between cellular dehydration and intracellular ice formation, with the cooling rate serving as the primary determinant of this equilibrium. Systematic research across diverse biological systems has consistently demonstrated that cooling rate optimization is essential for maintaining cell viability and tissue integrity. The interaction between cooling and warming rates further complicates this picture, requiring integrated protocol development. Future research directions should focus on cell-type specific parameter determination, improved predictive modeling of water transport, and the development of novel cryoprotective strategies that specifically target water kinetics pathways. The experimental methodologies outlined in this whitepaper provide a foundation for such investigations, enabling researchers to precisely quantify and manipulate water transport during freezing processes.
The survival of cells during cryopreservation is predominantly determined by the management of ice formation within a critical temperature window known as the Maximum Ice Formation Zone (MIFZ). Within this zone, the competing risks of intracellular ice formation (IIF) and excessive cellular dehydration reach their peak, making precise thermal control a paramount determinant of post-thaw viability. This whitepaper synthesizes current research on the biophysical processes governing ice formation in biological systems, examining how cooling rates, cryoprotective agent (CPA) concentration, and transfer temperatures interact within the MIFZ. Drawing upon recent experimental data and advanced modeling, we provide a technical framework for optimizing cryopreservation protocols to minimize cryoinjury for researchers and drug development professionals engaged in preserving cells, tissues, and advanced therapy medicinal products.
The Maximum Ice Formation Zone represents the temperature range during a freezing protocol where the majority of intracellular water undergoes phase change, either by crystallizing internally or by being transported out of the cell to freeze externally. The lower boundary of this zone is not defined by a single temperature but is influenced by cell type, CPA, and cooling rate. Navigating the MIFZ successfully requires a delicate balance: cooling too rapidly does not allow sufficient time for water to exit the cell, leading to lethal intracellular ice formation (IIF); cooling too slowly exposes the cell to prolonged solute toxicity and excessive dehydration, which can also cause damage [16] [17]. The central thesis of this whitepaper is that the MIFZ is the pivotal stage where the fate of a cryopreserved cell is decided, and its careful management is essential for optimizing recovery and function in research and drug development applications.
Core Biophysical Principles
The Competing Kinetics of Ice Formation and Cell Dehydration
During freezing, ice initially forms in the extracellular solution. This event creates an osmotic gradient that drives water out of the cell (cryodehydration), thereby concentrating the intracellular solutes. The rate of this water transport is a function of the membrane permeability to water and the temperature. Simultaneously, the supercooled cytoplasm becomes increasingly prone to intracellular ice nucleation. The probability of IIF is thus a race between the rate of water efflux (dehydration) and the cooling rate [17].
- Slow Cooling: Allows sufficient time for cellular dehydration, minimizing IIF but risking solute damage and excessive cell volume reduction.
- Rapid Cooling: Traps water inside the cell, leading to high supercooling and a high probability of nucleation and IIF, which is almost universally lethal [17].
Intracellular Ice Formation (IIF) Mechanisms
IIF is a nucleation-limited process that can occur via two primary mechanisms:
- Surface-Catalyzed Nucleation (SCN): Ice nucleates at the plasma membrane's inner surface and propagates into the cytoplasm.
- Volume-Catalyzed Nucleation (VCN): Ice nucleates spontaneously within the cytoplasm itself [17].
The improved probability of IIF (PIF) model incorporates a critical cell volume (Vf), which is the minimum volume a cell can achieve through dehydration. This model reveals that the maximum PIF during freezing may not reach 1 if the cell dehydrates to its critical volume before the temperature for nucleation is reached, a nuance not captured in earlier models [17].
The Role of Intercellular Ice Propagation in Tissues
In tissue constructs, IIF is not an isolated event per cell. Ice can propagate from one cell to its neighbors via gap junctions, significantly increasing the overall kinetics of ice formation within the tissue. A Markov chain model describing this process found that the total IIF rate in an unfrozen cell is the sum of the spontaneous IIF rate and the propagation rate from any adjacent frozen cells [18]. The nondimensional rate of intercellular ice propagation (α) has been measured, allowing for predictive modeling of IIF in larger tissue systems [18].
The Glass Transition and Vitrification
At ultra-low temperatures, highly concentrated cellular solutions can undergo a glass transition (vitrification), where the viscosity increases so dramatically that molecular diffusion effectively ceases, and the system becomes an amorphous solid without forming crystalline ice. This state is non-lethal. The temperature at which this intracellular colloidal glass transition (Tg'i) occurs, typically between -40°C and -60°C for many nucleated mammalian cells with CPAs, marks a safe endpoint for controlled slow cooling. Beyond this point, the risk of IIF is eliminated because the cellular contents are immobilized [16]. For free-living microbial cells in the presence of external ice, vitrification can occur at higher temperatures, between -10°C and -26°C, representing a fundamental lower thermal limit for metabolic activity [19].
Table 1: Key Biophysical Parameters and Their Impact within the MIFZ
| Parameter | Description | Impact of High Value | Impact of Low Value |
|---|---|---|---|
| Cooling Rate | Speed of temperature decrease through the MIFZ | Increases risk of intracellular ice formation (IIF) | Increases exposure to solute toxicity & dehydration |
| Membrane Permeability | Rate of water transport across the plasma membrane | Allows faster dehydration, reducing IIF risk | Slows dehydration, increasing IIF risk |
| CPA Concentration | Amount of penetrating cryoprotectant (e.g., DMSO) | Suppresses ice nucleation; increases toxicity | Reduces toxicity; increases IIF risk |
| Intracellular Tg'i | Temperature of intracellular colloidal glass transition | Higher safe transfer temperature to storage | Requires cooling to lower temperature for safety |
Experimental Data and Quantification
Determining the Critical Endpoint Temperature
Empirical studies have quantified the optimal endpoint temperature (ET) within the MIFZ, after which cells can be safely transferred to cryogenic storage. Research on nucleated mammalian cell lines (HepG2, CHO, MG63) demonstrated that controlling cooling to -40°C before plunging into liquid nitrogen ensured optimal post-thaw recovery, with no advantage gained by cooling to lower temperatures [16]. This ET aligns with DSC measurements showing an intracellular colloidal glass transition (Tg'i) for these cells occurring between -49°C and -59°C in the presence of DMSO. The ET of -40°C indicates that cells have dehydrated sufficiently to avoid significant damage upon transfer [16].
Table 2: Experimentally Determined Optimal Freezing Parameters for Various Systems
| System | Optimal Cooling Rate | Critical Endpoint Temperature | Key Metric | Source |
|---|---|---|---|---|
| HepG2, CHO, MG63 cells | 1°C/min | -40°C | Post-thaw cell recovery | [16] |
| C2C12 myoblasts | 1°C/min | -60°C (before fast cooling) | Cell viability (~65%) | [20] |
| Bovine oocytes | ~30,000°C/min | N/A (Ice-free via vitrification) | Ice formation (X-ray diffraction) | [21] |
| Strawberry tissue | 10.43 cm/h (fastest tested) | N/A | Water loss & cell structure preservation | [14] |
The Critical Impact of Warming
The MIFZ is critically relevant during both cooling and warming. For bovine oocytes, synchrotron-based X-ray diffraction revealed that even when cooling at ~30,000°C/min with CPA results in no detectable ice, a large fraction of ice consistent with the crystallization of most free water consistently forms during warming [21]. This finding underscores that most ice-related damage in such protocols must occur during the warming phase. The nature of the ice formed during cooling influences its behavior upon warming, and achieving much higher convective warming rates is a promising strategy to eliminate this damage [21].
The Influence of Freezing Rate on Microstructure
The physical morphology of the frozen state is directly controlled by the cooling rate. In frozen aqueous DMSO solutions, a slow cooling rate (1°C/min) leads to the formation of relatively large channels of freeze-concentrated solution (FCS), which effectively accommodate cells. In contrast, rapid cooling results in fine ice crystals and narrower FCS channels, which can inhibit cell accommodation and reduce recovery rates [20]. This principle is also observed in plant tissues; in strawberries, slow freezing rates cause significant water migration, large ice crystals, and severe damage to cell walls and membranes, whereas faster freezing better preserves the original cellular structure [14].
Essential Experimental Protocols
Protocol 1: Assessing Intracellular Ice Formation (IIF) in Cell Suspensions
This protocol utilizes a cryomicroscopy system to visualize IIF in real-time [17].
Detailed Methodology:
- Sample Preparation: Harvest and resuspend cells (e.g., HeLa cells) in an appropriate buffer (e.g., PBS) at a defined concentration.
- Loading: Pipette a 10 μL drop of cell suspension onto a coverslip and gently place a second coverslip on top, creating a thin film.
- Cooling and Seeding: Cool the sample on the cryostage from room temperature to -1°C at 10°C/min. Hold and seed extracellular ice by touching the solution edge with a pre-cooled copper wire. Warm to -0.5°C to minimize initial dehydration.
- Equilibration: Hold at -0.5°C for 3 minutes to allow for thermal and osmotic equilibration.
- Controlled Cooling: Cool the sample to a deep subzero temperature (e.g., -50°C) at various defined cooling rates (e.g., 5, 10, 15, 30, 60°C/min).
- Data Acquisition and Analysis:
- IIF Detection: Record video. IIF is identified by a sudden darkening or "flashing" of the cell cytoplasm.
- PIF Calculation: For each cooling rate, analyze a large number of cells (n ≥ 165) to determine the probability of IIF (PIF) as a function of temperature.
- Water Transport: Analyze cell volumetric changes from images to model water transport parameters.
Protocol 2: Determining the Optimal Endpoint Temperature via Cell Recovery
This protocol determines the temperature at which cells can be safely transferred to cryogenic storage [16].
Detailed Methodology:
- Cell Preparation: Culture cells (e.g., HepG2, CHO, MG63) to ~70% confluency. Trypsinize, centrifuge, and resuspend in complete culture medium.
- CPA Addition: Add DMSO in a single step to the cell suspension to a final concentration of 10% v/v and 10^6 cells/mL. Aliquot 1 mL into cryovials and equilibrate at 4°C.
- Controlled Rate Cooling: Cool the vials at 1°C/min in a controlled rate freezer from 4°C to various endpoint temperatures (ETs) between -10°C and -100°C.
- Transfer to Storage: At each designated ET, remove a set of vials (n=5) and plunge them directly into liquid nitrogen.
- Thawing and Assessment: Rapidly thaw the vials in a 37°C water bath. Assess post-thaw viability and function using:
- Viability Assay: Trypan blue exclusion to calculate the percentage of viable cells.
- Functional Assay: A metabolic activity assay (e.g., Cell Counting Kit-8) to measure recovery.
- Data Analysis: Plot post-thaw recovery against ET. The optimal ET is the temperature beyond which no further significant improvement in recovery is observed (e.g., -40°C).
The Researcher's Toolkit
Table 3: Essential Reagents and Instruments for MIFZ Research
| Item | Function/Description | Example Use Case |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating CPA; reduces IIF by lowering intracellular ice nucleation temperature. | Standard CPA for most mammalian cell lines, used at 5-10% v/v [20] [16]. |
| Controlled Rate Freezer | Apparatus that provides a precise, user-defined cooling rate. | Essential for slow cooling protocols (e.g., 1°C/min) through the MIFZ [16]. |
| Cryomicroscopy System | Microscope coupled with a temperature-controlled stage for visualizing ice formation in real-time. | Direct observation of IIF "flashing" and cell dehydration [17]. |
| Differential Scanning Calorimeter (DSC) | Measures thermal transitions, such as the glass transition (Tg'i), in a sample. | Determining the intracellular colloidal glass transition temperature to define a safe ET [16]. |
| Ice Recrystallization Inhibitors (IRIs) | Synthetic small molecules that inhibit the growth of ice crystals during warming. | Added to CPA cocktails to improve post-thaw recovery of sensitive cells like iPSCs and RBCs [22]. |
| Synchrotron X-ray Diffraction | High-intensity X-rays for quantitatively characterizing ice structure and volume within cells. | Detecting and quantifying ice volume fractions <1% in bovine oocytes during cooling/warming [21]. |
The Maximum Ice Formation Zone is the critical battlefield where the success of cryopreservation is determined. Mastering this zone requires a quantitative understanding of the interplay between cooling rate, CPA concentration, and the intrinsic biophysical properties of the cell. The experimental data and protocols outlined here provide a roadmap for researchers to define the MIFZ for their specific biological systems and to design protocols that steer cells toward survival.
Future advancements will likely focus on several key areas: the development of novel ice recrystallization inhibitors (IRIs) to mitigate warming damage [22], the refinement of ultra-rapid cooling and warming technologies to achieve ice-free cryopreservation with lower CPA toxicity [21] [23], and the creation of more sophisticated multi-scale models that predict IIF not just in single cells but in complex tissues [18]. By continuing to dissect the events within the MIFZ, the scientific community can develop more robust and reliable preservation methods, thereby accelerating progress in regenerative medicine, drug discovery, and biobanking.
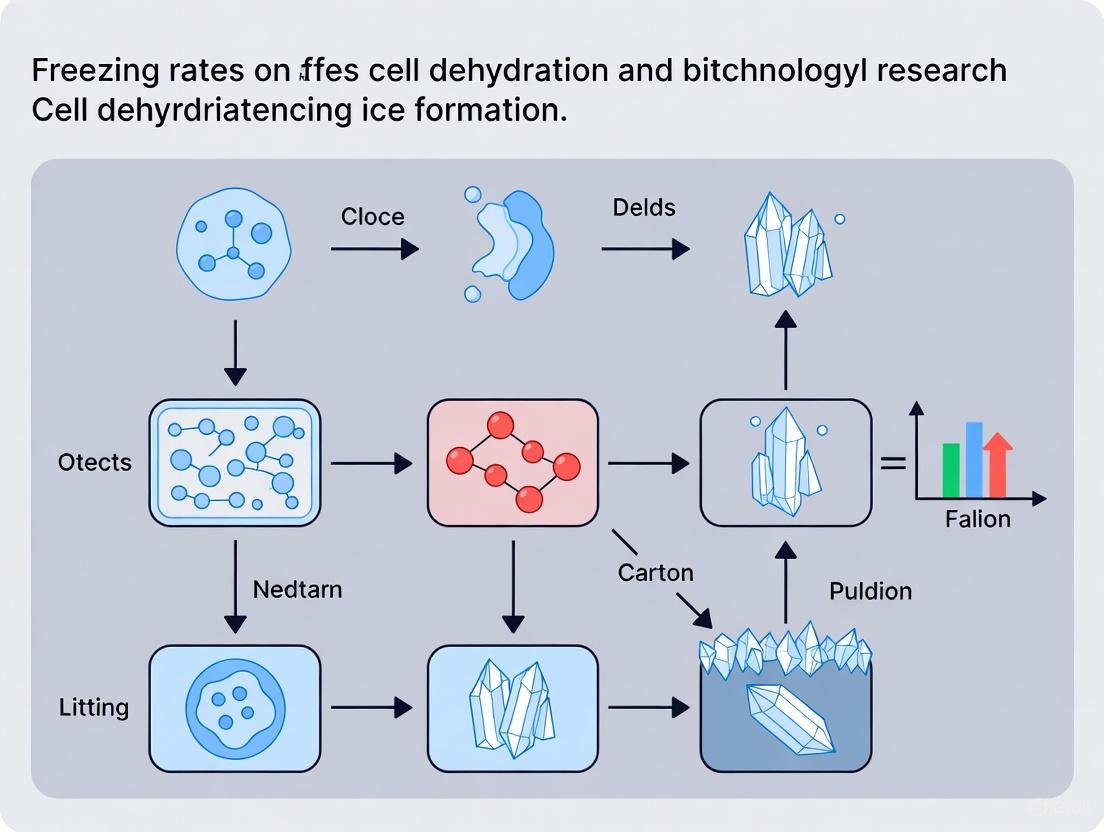
Cryopreservation is a cornerstone of modern biomedicine, enabling the long-term storage of cells for research, drug development, and clinical applications like cell and gene therapies [24]. The fundamental challenge it addresses is the mitigation of freezing stress, a phenomenon dominated by two primary, competing mechanisms: cell dehydration and ice formation [25] [26]. The severity of each is directly governed by the cooling rate. Slow cooling promotes excessive dehydration, causing "solution effects" from concentrated solutes, while rapid cooling leads to lethal intracellular ice formation (IIF) that disrupts cellular structures [25] [4]. Cryoprotectants (CPAs) are chemical agents designed to modulate these stresses. This whitepaper explores the mechanisms of the established CPA, dimethyl sulfoxide (DMSO), and evaluates the performance and action of emerging alternatives, framing the discussion within the critical context of freezing rate optimization to control cellular dehydration and ice formation.
Fundamental Mechanisms of Freezing Stress
Cellular damage during cryopreservation is a direct consequence of the physical and chemical changes water undergoes during freezing.
- Intracellular Ice Formation (IIF): At rapid cooling rates, intracellular water does not have sufficient time to exit the cell in response to extracellular ice formation. This trapped water then freezes internally, forming ice crystals that can puncture membranes and organelles, leading to immediate cell death [25] [26]. IIF is considered a primary cause of cryoinjury in fast-frozen samples.
- Cell Dehydration (The "Solution Effect"): At slow cooling rates, water gradually leaves the cell to equilibrate with the external osmotic pressure, leading to severe cell shrinkage. This concentrates intracellular salts and solutes to toxic levels, potentially denaturing proteins and disrupting lipid membranes [25] [4]. This "solution effect" is a major contributor to cell death in slowly frozen samples.
The freezing rate is therefore a critical parameter that determines which of these two damaging pathways predominates. Mazur's "two-factor hypothesis" establishes that an optimal cooling rate exists for each cell type, which balances the risks of IIF and excessive dehydration [25]. Recent research has highlighted that the ice nucleation temperature, the point at which ice first forms in the extracellular solution, is a key variable influencing this balance. Controlled ice nucleation at a higher temperature (e.g., -6°C) has been shown to promote more gradual dehydration and significantly reduce IIF in T cells, thereby improving post-thaw viability [27] [4].
DMSO: The Gold Standard and Its Limitations
Dimethyl sulfoxide (DMSO) is the most widely used penetrating cryoprotectant in clinical and research settings.
- Mechanism of Action: DMSO is a small, cell-penetrating molecule that protects cells through multiple mechanisms. It disrupts the hydrogen-bonding network of water, effectively lowering its freezing point and inhibiting ice crystal formation [28] [24]. By penetrating the cell, it reduces the amount of freezable water and mitigates the osmotic imbalance during freezing, thereby decreasing both IIF and solute damage [25] [4].
- Documented Limitations and Toxicity: Despite its efficacy, DMSO's use is associated with significant drawbacks. Clinically, infusion of DMSO-preserved cell therapies can cause adverse side effects, including cardiovascular, neurological, and allergic reactions [25] [28]. At a cellular level, DMSO can alter the expression of key markers on T and NK cells and impact their in vivo function and expansion potential [25]. Furthermore, its cytotoxicity can trigger apoptotic pathways through both mitochondrial and endoplasmic reticulum stress mechanisms [29].
Table 1: Quantitative Summary of DMSO's Role in Cryopreservation
| Aspect | Typical Concentration | Key Protective Function | Reported Drawbacks |
|---|---|---|---|
| Clinical Cell Therapy | 7–10% (v/v) [4] | Penetrates cell, prevents intracellular ice [4] | Clinical toxicities (allergic, cardiovascular) [25] |
| Research Grade | 5–15% (v/v) [30] | Lowers freezing point, stabilizes membranes [24] | Alters immune cell phenotype and function [25] |
| In Vitro Studies | 2.5–5% (v/v) [4] | Forms hydrogen bonds with water [28] | Induces apoptosis and necrosis [29] |
Emerging Alternatives and DMSO-Free Formulations
The documented limitations of DMSO have spurred intense research into safer and more specific alternatives. These can be broadly categorized as penetrating and non-penetrating CPAs.
- Sugars and Sugar-Based Derivatives: Trehalose is a non-penetrating disaccharide that protects cells via "water replacement," forming a glassy state (vitrification) that stabilizes membranes and proteins, and through "preferential exclusion" from protein surfaces, which promotes native protein folding [28]. Recent innovations involve engineering trehalose with functional groups from DMSO. For instance, sulfoxide-modified trehalose (Tre-S) was designed to mimic DMSO's hydrogen-bond disrupting ability while maintaining the biocompatibility of a sugar. This synthetic CPA has demonstrated an ability to inhibit ice formation and improve post-thaw survival of L929 cells, outperforming trehalose alone [28].
- Other Natural Cryoprotectants: Glycerol is another penetrating CPA with a long history of use. In bioink formulations for 3D cryobioprinting, 10% glycerol was found to improve viscosity and yield stress, leading to significantly better cell viability after cryopreservation at -80°C compared to DMSO-containing or CPA-free bioinks [30]. Additionally, recombinant plant proteins, such as wheat enolase (TaENO) and dehydrin (WCS120), have shown remarkable cryoprotective efficacy for rat hepatocytes. These proteins function by inhibiting ice recrystallization and, crucially, by suppressing the activation of mitochondrial and endoplasmic reticulum stress-induced apoptosis that is typically triggered by DMSO [29].
- Polymers and Zwitterions: Synthetic polymers like hydroxyethyl starch and polyvinyl pyrrolidone act as non-penetrating CPAs. They protect cells by increasing solution viscosity, modifying ice crystal growth, and reducing the osmotic shock during the addition and removal of CPAs [25] [30]. Zwitterionic molecules, which contain both positive and negative charges, have also been explored for their ability to mimic antifreeze proteins and stabilize cell membranes without toxicity [28].
Table 2: Comparison of Emerging DMSO-Free Cryoprotectants
| Cryoprotectant | Type | Proposed Mechanism of Action | Reported Cell Type / Model |
|---|---|---|---|
| Sulfoxide-modified Trehalose (Tre-S) | Synthetic Penetrating | Disrupts water H-bond network; balances osmotic pressure [28] | L929 Fibroblasts [28] |
| Glycerol | Penetrating | Interacts with water & bioink polymers; reduces ice formation [30] | Human Dermal Fibroblasts (in bioink) [30] |
| Recombinant Plant Proteins (e.g., TaENO, WCS120) | Non-Penetrating | Inhibits ice recrystallization; attenuates apoptosis pathways [29] | Rat Hepatocytes [29] |
| Hydroxyethyl Starch | Non-Penetrating Polymer | Increases viscosity; reduces osmotic stress [25] [30] | NK cells, T cells [25] |
Experimental Protocols for Evaluating Cryoprotectants
A robust evaluation of novel CPAs requires a multi-faceted approach, correlating post-thaw viability with direct observation of freezing dynamics.
Protocol 1: Bulk Freeze-Thaw Viability Assessment This standard protocol assesses the functional outcome of cryopreservation.
- Cell Preparation: Culture the model cell line (e.g., Jurkat T cells) to a log-phase density of 1x10^6 cells/mL [4].
- CPA Addition: Resuspend the cell pellet in the test cryoformulation (e.g., 5-10% DMSO in Plasma-Lyte A, or a DMSO-free alternative). Use a non-CPA medium as a negative control.
- Freezing: Aliquot the cell suspension into cryovials. Use a controlled-rate freezer, cooling at -1°C/min from 4°C to a set point between -40°C and -80°C, before transferring to liquid nitrogen for storage [25] [4]. For controlled ice nucleation studies, initiate nucleation at a specific temperature (e.g., -6°C) using a pressure shift or other nucleation device [27] [4].
- Thawing: Rapidly thaw the vials in a 37°C water bath until the last ice crystal disappears [24].
- Viability Analysis: Dilute the thawed sample gradually with culture medium to minimize osmotic shock. Assess cell membrane integrity using dye exclusion (e.g., Trypan Blue) and/or flow cytometry with propidium iodide and acridine orange [4]. For functional assessment, culture the recovered cells and measure metabolic activity, proliferation, and phenotype over several days [25].
Protocol 2: Cryomicroscopic Analysis of Intracellular Events This protocol provides direct, mechanistic insight into how CPAs and freezing rates modulate cellular responses [27] [4] [26].
- Sample Loading: Place a small droplet (~5 µL) of cell suspension in the test cryoformulation on a specialized cryomicroscopy stage.
- Freezing and Imaging: Program the stage to replicate controlled freezing rates (e.g., -1°C/min to -10°C/min). Use polarized light to visualize extracellular ice formation and a fluorescent DNA stain (e.g., propidium iodide) to monitor membrane integrity in real-time.
- Data Collection: Record the temperature of ice nucleation. Track changes in cell volume to quantify dehydration. Identify the onset of IIF, which is indicated by a sudden darkening of the cytoplasm [4] [26].
- Correlation: Correlate the recorded intracellular events (dehydration extent, IIF occurrence) with the post-thaw viability data from Protocol 1 to establish a mechanism-action relationship for the CPA.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Cryopreservation Research
| Reagent / Solution | Function in Research |
|---|---|
| Dimethyl Sulfoxide (DMSO) | Benchmark penetrating CPA; positive control for evaluating new formulations [25] [4]. |
| Trehalose and Sucrose | Non-penetrating CPAs; often used in combination with other agents to augment vitrification and stabilize membranes [28]. |
| Plasma-Lyte A | A balanced salt solution; commonly used as the base solution for clinical-relevant cryoformulations [27] [4]. |
| Propidium Iodide / Acridine Orange | Fluorescent viability stains used in cryomicroscopy and flow cytometry to distinguish live/dead cells based on membrane integrity [4]. |
| Controlled-Rate Freezer | Equipment that ensures a reproducible, optimized cooling profile, which is critical for standardizing experiments and scaling up to manufacturing [4] [24]. |
The role of cryoprotectants in modulating freezing stress is complex and inextricably linked to the physics of ice formation and cell dehydration. While DMSO remains the most effective and widely used CPA, its clinical and cellular toxicities are a significant driver for innovation. The future of cryopreservation lies in the rational design of DMSO-free solutions, such as engineered biomimetic molecules (e.g., Tre-S) and natural protein-based protectants, which target specific pathways of cryoinjury and apoptosis. Success in this field requires an integrated approach that combines a deep understanding of freezing rate dynamics, direct observation of cellular events, and a rigorous assessment of post-thaw cell function. As cell and gene therapies continue to advance, the development of safer, more effective cryoprotectants will be paramount to ensuring the global deployment of viable and potent cellular products.
Visualizing Pathways and Workflows
The following diagrams summarize the key concepts and experimental workflows discussed in this whitepaper.
Diagram 1: Freezing stress pathways.
Diagram 2: Cryoprotectant mechanisms.
The freezing of water is a fundamental process with profound implications in fields ranging from climate science to the preservation of biological cells. At the heart of understanding this phenomenon lies Classical Nucleation Theory (CNT), the primary theoretical framework used to quantitatively describe the kinetics of phase transitions, including the formation of ice from supercooled water [31] [32]. Nucleation is the critical first step in the spontaneous formation of a new thermodynamic phase from a metastable state, and the time required for nucleation can vary by orders of magnitude, ultimately determining how long it takes for the new phase to appear [31].
Within the context of cryopreservation research for drug development and cell therapies, the principles of CNT are indispensable. The formation of ice crystals during freezing can mechanically disrupt cellular membranes and cause lethal increases in solute concentration, leading to cell death [10]. Therefore, understanding and controlling ice nucleation is not merely an academic exercise but a practical necessity for improving the viability of cryopreserved cells, tissues, and therapeutic products [33] [4]. This article provides an in-depth examination of CNT, explores its application in controlling ice formation for cell preservation, and details relevant experimental methodologies.
Theoretical Foundations of Classical Nucleation Theory
Classical Nucleation Theory describes the formation of a new phase as an activated process that requires the system to surmount a free energy barrier. This barrier arises from the competition between the bulk free energy of the new phase and the interfacial free energy created by forming a boundary between the new and old phases [31] [32].
The Free Energy of Nucleation
For the homogeneous formation of a spherical ice nucleus within supercooled water, the change in Gibbs free energy, ΔG, is given by:
ΔG = (4/3)πr³Δgv + 4πr²σ
Here, r is the radius of the nucleus, Δg<sub>v</sub> is the volumetric Gibbs free energy difference between the ice and water phases (which is negative below the melting point), and σ is the specific interfacial free energy (surface tension) between the ice and water [31].
The first term, the volume term, represents the energy gain from forming the more stable ice phase. The second term, the surface term, represents the energy cost of creating the new ice-water interface. For small values of r, the positive surface term dominates, making ΔG positive. As r increases, the negative volume term, which scales with r³, begins to dominate over the positive surface term, which scales with r² [31]. This relationship results in a free energy profile that initially increases with nucleus size, reaches a maximum, and then decreases.
Critical Nucleus and Activation Barrier
The size at which the free energy is maximized is known as the critical radius, r. Nuclei smaller than r* are unstable and tend to dissolve, while those larger than r* are stable and will grow spontaneously [31]. The critical radius and the height of the activation barrier, ΔG, can be derived by setting the derivative of ΔG with respect to r equal to zero:
r* = -2σ / Δgv
ΔG* = (16πσ³) / (3|Δgv|²)
The volumetric free energy difference Δg<sub>v</sub> becomes more negative as the degree of supercooling (the difference between the melting point and the actual temperature) increases. Consequently, both the critical radius r* and the activation barrier ΔG* decrease with increasing supercooling [31]. This is a key insight, as it explains why nucleation becomes more probable at lower temperatures.
Homogeneous vs. Heterogeneous Nucleation
The theory described above pertains to homogeneous nucleation, where ice nuclei form spontaneously and randomly within the bulk water. However, this is a rare event requiring significant supercooling [31]. In practice, heterogeneous nucleation is far more common, occurring on surfaces, impurities, or container walls [31].
CNT accounts for heterogeneous nucleation by introducing a geometric factor f(θ) that depends on the contact angle θ between the ice nucleus and the substrate. This factor reduces the activation barrier:
ΔGhet = f(θ) ΔGhom
where f(θ) = (2 - 3cosθ + cos³θ)/4 [31]. For a surface that perfectly wets the nucleus (θ=0°), the barrier is eliminated, while for a completely non-wetting surface (θ=180°), the barrier is identical to homogeneous nucleation. In real-world scenarios, including cryopreservation, the presence of various surfaces and impurities means heterogeneous nucleation dominates and occurs at higher, more biologically relevant temperatures [31] [32].
The Nucleation Rate
The central quantitative prediction of CNT is the nucleation rate, R, which represents the number of nucleation events per unit volume per unit time. It is given by an Arrhenius-type equation:
R = NS Z j exp(-ΔG* / kBT)
- NS: The number of potential nucleation sites per unit volume.
- Z: The Zeldovich factor, a kinetic factor related to the probability that a critical nucleus will continue to grow rather than dissolve.
- j: The rate at which molecules attach to the critical nucleus.
- ΔG*: The free energy barrier for forming a critical nucleus.
- kB: Boltzmann's constant.
- T: Temperature [31].
The exponential term dominates the temperature dependence of the rate, leading to the immense variation observed in nucleation times [31].
Table 1: Key Parameters in Classical Nucleation Theory
| Parameter | Symbol | Description | Role in CNT |
|---|---|---|---|
| Critical Radius | r* |
The minimum stable nucleus size. | Nuclei smaller than r* dissolve; larger ones grow. |
| Activation Barrier | ΔG* |
The free energy maximum that must be overcome. | Determines the exponential probability of nucleation. |
| Interfacial Energy | σ |
The free energy per unit area of the ice-water interface. | The source of the activation barrier; a higher σ increases ΔG*. |
| Volumetric Free Energy | Δg<sub>v</sub> |
The free energy difference per unit volume between ice and water. | The driving force for nucleation; becomes more negative with supercooling. |
| Nucleation Rate | R |
The number of nucleation events per unit volume per unit time. | The central output of CNT, predicting how fast ice forms. |
CNT in the Context of Cryopreservation and Cell Viability
The principles of CNT directly inform the challenges and strategies of cryopreservation. The goal is to navigate the competing damage mechanisms of intracellular ice formation (IIF) and excessive dehydration by controlling the nucleation and growth of ice [4].
The Dual Threats of Freezing to Cells
When an aqueous cell suspension is cooled, water first freezes extracellularly. This event has two major consequences for cells:
- Solute Concentration (Osmotic Stress): As pure water freezes, solutes are excluded from the ice lattice and become concentrated in the remaining liquid phase. This creates a hypertonic extracellular environment, driving water out of the cell through osmosis. Excessive dehydration can lead to damaging increases in intracellular solute concentration and cell shrinkage [10] [4].
- Intracellular Ice Formation (IIF): If the cooling rate is too rapid, there is insufficient time for water to leave the cell. The supercooled intracellular water may then nucleate, forming ice crystals inside the cell. IIF is almost always lethal, as it can mechanically disrupt cellular membranes and organelles [10] [4].
A slow cooling rate (e.g., -1 °C/min) is often used for mammalian cells to allow time for osmotic dehydration and to avoid IIF. Conversely, rapid warming during thawing is preferred to minimize the growth of damaging ice crystals through a process known as recrystallization [4].
The Critical Role of Ice Nucleation Temperature
The temperature at which extracellular ice nucleates is a critical variable. In uncontrolled freezing, nucleation occurs spontaneously at a variable and unpredictable degree of supercooling. According to CNT, a lower nucleation temperature (higher supercooling) results in a lower activation barrier and a higher nucleation rate, leading to the formation of a larger number of small ice crystals [20].
Controlled Ice Nucleation (CIN) is a technique where the nucleation event is triggered at a specific, higher temperature (e.g., -5 °C instead of the spontaneous -10 °C or lower). This is achieved through methods like manual ice seeding, shock cooling, or pressure shift [4]. The impact of this control is significant:
- Larger Ice Crystal Morphology: Nucleating at a higher temperature (less supercooling) results in fewer, larger ice crystals. Research on frozen dimethyl sulfoxide (DMSO) solutions has shown that a slow cooling rate of 1 °C/min produces relatively large, channel-like networks of freeze-concentrated solution (FCS), where cells can be accommodated [20].
- Reduced Intracellular Ice Formation: Studies on Jurkat T-cells have demonstrated that initiating ice formation at a warmer temperature (e.g., -6 °C) promotes greater cellular dehydration during freezing, thereby reducing the incidence of lethal IIF [4].
- Improved Process Consistency: CIN ensures a consistent freezing history across all samples in a batch, reducing heterogeneity in post-thaw cell recovery and product quality [4].
Table 2: Impact of Cooling Rate and Nucleation on Cryopreservation Outcomes
| Parameter | Slow Cooling (~1°C/min) | Rapid Cooling (>10°C/min) | Controlled Nucleation |
|---|---|---|---|
| Extracellular Ice | Forms larger, channel-like FCS networks [20]. | Forms numerous, fine ice crystals with narrow FCS [20]. | Initiates at a defined, warmer temperature. |
| Cell Dehydration | Allows time for sufficient water efflux [4]. | Insufficient time for dehydration, high risk of IIF [4]. | Promotes greater dehydration, reducing IIF risk [4]. |
| Typical Cell Viability | Higher recovery (e.g., 65% for C2C12 myoblasts) [20]. | Lower recovery (e.g., 54% for C2C12 myoblasts) [20]. | Can improve recovery and reduce required CPA concentration [4]. |
Experimental Protocols for Studying Ice Nucleation
Translating the theory of CNT into practical cryopreservation protocols requires robust experimental methods to observe and quantify ice formation and its effects on cells.
Microscopic Observation of Freeze-Concentrated Solutions (FCS)
This protocol is used to visualize the morphology of ice crystals and the surrounding FCS channels under different freezing conditions [20].
Methodology:
- Sample Preparation: A 10 μL aliquot of a cryoprotectant solution (e.g., DMSO in water at 5-20 wt%) is stained with a fluorescent dye like sodium fluorescein (100 μM) and sandwiched between two slide glasses.
- Freezing Stage: The sample is placed on a temperature-controlled cooling stage mounted on an upright fluorescent microscope.
- Controlled Freezing: The solution is cooled at a defined rate (e.g., 1 °C/min, 10 °C/min, 30 °C/min) to a target temperature (e.g., -60 °C).
- Image Acquisition: A CMOS camera is used to capture fluorescence images during the freezing process. The FCS, where the solute and dye are concentrated, appears bright, while the pure ice crystals appear dark.
- Image Analysis: Software such as ImageJ is used to measure the width of the FCS channels and the size of the ice particles statistically.
Key Findings: Using this method, researchers confirmed that a slow cooling rate of 1 °C/min produces larger FCS channels, which are more effective at accommodating cells and are associated with higher post-thaw viability [20].
Investigating Intracellular Dehydration and IIF in T-Cells
This protocol leverages a thin-film microscopy apparatus to directly monitor cellular responses in real-time during freezing [4].
Methodology:
- Cell Preparation: Jurkat T-cells (a model for CAR-T cells) are suspended in cryoformulations with varying DMSO concentrations (e.g., 2.5%, 5%, and 10% v/v in Plasma-Lyte A).
- Thin-Film Setup: A small volume of cell suspension is created as a thin film on a temperature-controlled microscope stage.
- Controlled Nucleation: Ice nucleation is triggered at specific temperatures (e.g., -6 °C and -10 °C) using a controlled method, and compared to spontaneous nucleation.
- Multi-Modal Imaging:
- Polarized Light: Used to identify the formation of ice crystals.
- Fluorescence Microscopy: Cells are stained with viability dyes (e.g., acridine orange and propidium iodide) to monitor membrane integrity and cell volume changes in real-time. Darkening of cellular appearance is interpreted as intracellular ice formation (IIF).
- Data Correlation: The timing and extent of cellular dehydration and IIF are correlated with the final post-thaw viability, measured using standard assays like trypan blue exclusion.
Key Findings: This approach revealed that controlled nucleation at -6 °C, closer to the equilibrium freezing point, promoted better cellular dehydration and resulted in less IIF compared to spontaneous nucleation at lower temperatures [4].
The Scientist's Toolkit: Essential Reagents and Materials
Successful cryopreservation relies on a combination of specialized equipment and chemical agents designed to modulate ice nucleation and protect cells.
Table 3: Key Research Reagent Solutions for Cryopreservation Studies
| Reagent / Material | Function / Role | Example Application |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | A permeating cryoprotectant (CPA). Penetrates the cell, depresses the freezing point, reduces IIF by forming hydrogen bonds with water, and increases membrane porosity [10] [4]. | Used at 5-10% (v/v) as a standard CPA for freezing mammalian cells like T-cells and stem cells [10] [4]. |
| Trehalose | A non-permeating disaccharide CPA. Protects cells by stabilizing membranes and proteins, often through water substitution and vitrification mechanisms [10] [34]. | Used in combination with other CPAs (e.g., skim milk) to improve survival of probiotics like Lactobacillus during freeze-drying [34]. |
| Skim Milk | A non-permeating CPA. Forms a rigid, viscous matrix that inhibits ice crystal growth and provides a protective microencapsulation for cells [34]. | Serves as a stabilizing matrix for bacteria during freeze-drying; often used with trehalose [34]. |
| Polyethylene Glycol (PEG) | A non-permeating polymeric CPA. Induces vitrification and can inhibit ice recrystallization during thawing [33]. | Used in vitrification mixtures and DMSO-free cryoformulations [33]. |
| Controlled Rate Freezer | Equipment that provides a precise, user-defined cooling profile (e.g., 1°C/min) to optimize dehydration and minimize IIF [4]. | Essential for the reproducible freezing of sensitive cell types like CAR-T cells and stem cells [20] [4]. |
| Ice Nucleators | Agents or devices used to trigger controlled ice nucleation at a defined temperature, ensuring process consistency [4]. | Methods include pressure shift (Control Lyo), chemical nucleators, or manual ice seeding [4]. |
Classical Nucleation Theory provides the fundamental physical principles that govern the initiation of ice formation. Its concepts of critical radius, activation barrier, and nucleation rate are critical for understanding and predicting the behavior of water during supercooling. In the context of preserving biological cells, CNT directly explains the profound impact that cooling rates and nucleation temperatures have on ice crystal morphology, intracellular dehydration, and ultimately, cell survival.
The experimental evidence is clear: moving from uncontrolled, spontaneous nucleation to controlled ice nucleation at warmer temperatures can create a more favorable freezing environment, promoting the formation of less damaging ice structures and enhancing cell dehydration to avoid lethal intracellular ice formation. As cell therapies and regenerative medicine continue to advance, the precise application of CNT, combined with advanced cryoprotectant strategies and standardized freezing protocols, will be essential for ensuring the high viability, functionality, and consistency of these vital medical products. Future research will continue to refine these protocols and explore the potential of novel ice-binding materials and DMSO-free cryoformulations to further improve cryopreservation outcomes [33].
From Theory to Practice: Methodologies for Controlling Freezing Rates and Ice Nucleation
The preservation of biological materials at low temperatures is a cornerstone of modern biomedical research, clinical applications, and drug development. The spectrum of freezing techniques—encompassing slow freezing, rapid freezing, and vitrification—represents a critical technological arsenal for maintaining cellular viability and function during long-term storage. These techniques fundamentally aim to mitigate the two primary mechanisms of cryoinjury: ice crystal formation and deleterious cellular dehydration [35].
The core challenge in cryopreservation lies in navigating the physical phase changes of water. When biological systems cool below their freezing point, intracellular and extracellular water can form ice crystals that mechanically disrupt membranes and subcellular structures [13]. Concurrently, the conversion of water to ice concentrates dissolved solutes, leading to osmotic stress and biochemical damage [1]. The efficacy of any freezing protocol is therefore determined by its ability to control these competing damaging processes, largely through the precise manipulation of cooling rates and the use of cryoprotective agents (CPAs) [36].
This technical guide provides an in-depth examination of how different cooling rates across the freezing spectrum influence the critical balance between cellular dehydration and ice formation. It is structured to serve researchers, scientists, and drug development professionals by integrating fundamental principles, quantitative data comparisons, detailed experimental protocols, and practical toolkits for implementation.
Fundamental Principles and Definitions
Technical Definitions and Characteristics
Slow Freezing: A process involving a gradual, controlled reduction in temperature, typically at rates of -0.1°C/min to -3°C/min [37]. This technique allows sufficient time for extracellular ice formation and subsequent osmotic efflux of intracellular water, thereby minimizing lethal intracellular ice crystallization [36]. However, it exposes cells to prolonged hypertonic stress and potential "solute effects" from concentrated electrolytes.
Rapid Freezing: Characterized by substantially higher cooling rates, often exceeding -2500°C/min [37]. This approach minimizes time for cellular dehydration but significantly increases the risk of intracellular ice formation (IIF) as water molecules do not have sufficient time to exit the cell before reaching temperatures that facilitate ice nucleation [4].
Vitrification: An ultra-rapid cooling process that achieves a non-crystalline, glass-like solid state through exceptionally high cooling rates (often greater than -10,000°C/min) combined with high concentrations of CPAs [37]. This method completely avoids ice crystallization by solidifying the solution into an amorphous glass, thereby circumventing both mechanical damage from ice and excessive dehydration [36].
The Underlying Physics: Cellular Dehydration and Ice Formation
The interplay between cooling rate and cellular response is elegantly explained by Mazur's "two-factor hypothesis" [1]. During freezing, extracellular water freezes first, creating a chemical potential gradient that drives intracellular water outward through osmosis. The success of this process depends critically on the cooling rate:
- At slow cooling rates, water has adequate time to exit the cell, resulting in protective dehydration but potentially harmful solute concentration.
- At rapid cooling rates, water cannot exit quickly enough, leading to supercooling and eventual intracellular ice formation.
- Vitrification represents the optimal compromise, using ultra-rapid cooling and CPAs to prevent both excessive dehydration and ice formation.
The following diagram illustrates how different cooling rates affect the balance between cellular dehydration and ice formation, determining the success of cryopreservation:
Table 1: Comparative Analysis of Freezing Techniques
| Parameter | Slow Freezing | Rapid Freezing | Vitrification |
|---|---|---|---|
| Cooling Rate | -0.1°C to -3°C/min [37] | >-2,500°C/min [37] | >-10,000°C/min to -50,000°C/min [37] |
| CPA Concentration | Low (1-2 M) [38] | Moderate | High (4-8 M) [36] |
| Primary Ice Formation | Extracellular [37] | Intra- and extracellular | None (amorphous glass) [36] |
| Cellular Dehydration | Significant [1] | Minimal | Moderate (pre-equilibrated) |
| Intracellular Ice | Minimal [36] | Extensive [4] | None |
| Primary Damage Mechanisms | Solute effects, osmotic shock [1] | Intracellular ice mechanical damage [4] | CPA toxicity, glass fracture [36] |
| Optimal Warming Rate | Slow to moderate | Rapid | Ultra-rapid (prevents devitrification) [37] |
Experimental Methodologies and Protocols
Protocol for Slow Freezing of Ovarian Tissue
The slow freezing protocol for ovarian tissue cryopreservation represents a well-established methodology for fertility preservation [39]:
- Tissue Preparation: Dissect ovarian tissue into pieces approximately 10×10×1-2 mm using a scalpel under sterile conditions.
- CPA Equilibration: Transfer tissue to freezing medium containing L-15 medium supplemented with 10% Serum Substitute Supplement (SSS), 10% DMSO, and 0.1 M sucrose. Equilibrate for 30 minutes at 4°C.
- Programmed Freezing:
- Load tissue into cryovials and place in programmable freezer.
- Cool from 2°C to -6°C at -2°C/min.
- Initiate ice seeding at -6°C.
- Cool to -40°C at -0.3°C/min.
- Rapid cool to -140°C at -10°C/min.
- Storage: Transfer samples to liquid nitrogen for long-term storage at -196°C.
- Thawing: Warm cryovials in 37°C water bath for 2 minutes, then gradually remove CPAs using decreasing sucrose concentrations (0.5 M, 0.25 M, 0.125 M, 0 M) with 10% SSS.
Protocol for Vitrification of Oocytes/Embryos
The vitrification process for gametes and embryos requires precise timing and specialized devices [37] [39]:
- Equilibration: Expose oocytes/embryos to equilibration solution (e.g., 7.5% ethylene glycol + 7.5% DMSO) for 10-15 minutes at room temperature.
- Vitrification Solution: Transfer to vitrification solution (e.g., 15% ethylene glycol + 15% DMSO + 0.5 M sucrose) for 60 seconds at room temperature.
- Loading and Cooling:
- Place cells in minimal volume (<1 μL) on cryodevice (e.g., Cryotop, Open Pulled Straw).
- Immediately plunge into liquid nitrogen within 60 seconds of vitrification solution exposure.
- Storage: Maintain at -196°C in liquid nitrogen.
- Warming:
- Rapidly warm by transferring directly into warming solution at 37°C.
- Sequentially transfer through decreasing sucrose concentrations (0.5 M, 0.25 M, 0.125 M, 0 M) to remove CPAs.
Experimental Setup for Monitoring Intracellular Ice Formation
Advanced microscopy techniques enable real-time observation of ice formation dynamics [4]:
- Apparatus Setup: Utilize an upright fluorescent microscope equipped with a temperature-controlled cooling stage and high-speed camera.
- Sample Preparation: Sandwich 10 μL cell suspension between two slide glasses. For fluorescence imaging, incorporate membrane-impermeable dyes (e.g., propidium iodide) to monitor membrane integrity.
- Temperature Programming: Cool samples at controlled rates (1°C/min to 50°C/min) while monitoring phase transitions.
- Image Analysis:
- Use polarized light to identify ice crystal formation.
- Employ fluorescence to track cell volume changes and membrane integrity.
- Quantify intracellular ice formation by detecting sudden darkening of cellular appearance.
- Data Collection: Document the temperature of intracellular ice formation, ice crystal morphology, and correlation with post-thaw viability.
Comparative Performance Data
Quantitative Outcomes Across Cell and Tissue Types
Table 2: Experimental Outcomes Across Freezing Techniques
| Cell/Tissue Type | Freezing Method | Cooling Rate | Viability/Outcome | Reference |
|---|---|---|---|---|
| Ovarian Tissue Follicles | Slow Freezing | ~ -0.3°C/min | 96% viability (RR=0.96) | [38] |
| Ovarian Tissue Follicles | Vitrification | > -10,000°C/min | No significant difference vs. slow freezing | [38] |
| C2C12 Myoblasts | Slow Freezing | -1°C/min | 65% recovery | [40] |
| C2C12 Myoblasts | Rapid Freezing | -30°C/min | 54% recovery | [40] |
| Jurkat (T-) Cells | Controlled Nucleation | -1°C/min | Enhanced dehydration, reduced IIF | [4] |
| Human Oocytes | Vitrification (Cryotop) | > -10,000°C/min | >90% survival | [37] |
| CHO Cells | Vitrification (No CPA) | -120°C/min | <10% viability | [1] |
Impact on Specific Cellular Components
The structural integrity of cellular components varies significantly across freezing techniques:
- Stromal Cells: Ovarian tissue stromal cells showed higher apoptosis in slow freezing (SF) compared to vitrification at 4 weeks post-transplantation, though this difference normalized by 6 weeks [39].
- Blood Vessel Integrity: CD31 expression (marker of angiogenesis) was better preserved in slow-frozen tissues compared to vitrified ones after transplantation [39].
- DNA Integrity: The proportion of DNA fragmented follicles showed no significant difference between slow freezing and vitrification (RR=1.20) [38].
- Membrane Integrity: Rapid freezing caused substantial membrane damage in Jurkat cells, while controlled nucleation at -6°C improved membrane preservation [4].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents and Equipment for Freezing Technique Research
| Category | Specific Examples | Function/Application | Notes |
|---|---|---|---|
| Permeable CPAs | DMSO, Ethylene Glycol (EG), Glycerol | Penetrate cell membrane; reduce intracellular ice formation [37] | DMSO concentration typically 1.0-1.5 M for slow freezing; 4-6 M for vitrification [35] |
| Non-Permeable CPAs | Sucrose, Trehalose, Ficoll | Provide extracellular protection; control osmotic pressure [1] | Sucrose commonly used at 0.1-0.5 M concentrations [39] |
| Ice Regulators | Antifreeze Proteins (AFPs), Polyvinyl Alcohol | Control ice nucleation and recrystallization [35] | Synthetic mimics being developed to reduce costs |
| Cryodevices | Cryotop, Open Pulled Straw (OPS), Cryoloop | Enable ultra-rapid cooling for vitrification [37] | Open systems allow faster cooling but risk liquid nitrogen contamination |
| Specialized Equipment | Programmable Freezer, Cooling Stage Microscope | Precise rate control; real-time observation [4] [39] | Essential for protocol standardization |
| Viability Assays | Trypan Blue Exclusion, CCK-8 Kit, TUNEL Assay | Assess post-thaw cell recovery and function [39] [40] | Multiple assays recommended for comprehensive assessment |
Advanced Concepts and Emerging Technologies
Novel Approaches to Ice Control
Controlled Ice Nucleation: Precisely initiating extracellular ice formation at defined temperatures (e.g., -6°C instead of spontaneous nucleation at lower temperatures) promotes gradual cellular dehydration and significantly reduces intracellular ice formation in T-cells [4].
Aquaporin Enhancement: Engineering cells to overexpress water channel proteins (e.g., AQP4) increases membrane water permeability, enabling faster dehydration during cooling and improved survival at rapid cooling rates [1].
Ice-Blocking Polymers: Synthetic and natural polymers, including poly(vinyl alcohol) and antifreeze protein mimics, can inhibit ice recrystallization during thawing, reducing mechanical damage to cell membranes [35].
Nanotechnology Approaches: Nanoparticles and hydrogels provide scaffolding that controls ice crystal growth and distribution, particularly valuable for preserving more complex tissues and multicellular systems [35].
The Critical Role of Warming Rates
The warming rate is equally crucial, and in some cases more important, than the cooling rate for successful cryopreservation:
- Devitrification: During warming of vitrified samples, ice crystallization can occur if warming is too slow, defeating the purpose of vitrification [36].
- Recrystallization: In slowly frozen samples, small ice crystals merge into larger, more damaging structures during slow warming [13].
- Optimal Protocols: Ultra-rapid warming (>500°C/min) is typically required for vitrified samples, while moderate rates are sufficient for slowly frozen cells [37].
The following workflow summarizes the decision process for selecting and optimizing a cryopreservation protocol based on cell type and application requirements:
The spectrum of freezing techniques offers researchers a diverse toolkit for preserving biological materials, with each approach presenting distinct advantages and limitations rooted in their fundamental effects on cellular dehydration and ice formation. Slow freezing promotes protective dehydration but risks solute damage; rapid freezing minimizes dehydration but promotes intracellular ice formation; vitrification avoids ice crystals but requires toxic CPA concentrations.
The optimal technique depends critically on the specific biological system, with factors including cell volume, membrane permeability, natural cryotolerance, and intended post-thaw application dictating the most appropriate approach. Emerging technologies—particularly controlled ice nucleation, molecular ice regulators, and membrane engineering—promise to enhance cryopreservation outcomes across all techniques.
Future advancements will likely focus on developing less toxic CPAs, improving warming protocols, and creating customized approaches for complex tissues and organs. As cryopreservation continues to evolve, the precise control of freezing rates will remain central to balancing cellular dehydration and ice formation, enabling more effective preservation of increasingly diverse biological systems for research and clinical applications.
Controlled-rate freezing is an indispensable technological process in modern biomedical research and therapy development, serving as a cornerstone for preserving cellular integrity, function, and viability. This technique systematically manages the cooling rate of biological specimens to mitigate the two primary mechanisms of cryoinjury: intracellular ice formation and excessive cellular dehydration. For sensitive cell types—including therapeutic T cells, stem cells, and other primary cells—the precision of this process becomes particularly critical as even minor deviations can compromise cellular function and viability. The fundamental challenge lies in navigating the physical processes that occur during freezing: as extracellular water crystallizes, solute concentration increases osmotically, drawing water out of cells. If cooling proceeds too rapidly, insufficient dehydration occurs, resulting in lethal intracellular ice. Conversely, excessively slow cooling exposes cells to prolonged hypertonic stress and cryoprotectant toxicity [4].
This technical guide examines standard controlled-rate freezing protocols within the context of contemporary research on cellular dehydration and ice formation dynamics. The protocols outlined herein are specifically designed for researchers, scientists, and drug development professionals working with sensitive cell types, providing both theoretical foundations and practical methodologies to optimize cryopreservation outcomes. By understanding and controlling the interplay between cooling rates and cellular responses, practitioners can significantly enhance post-thaw recovery, maintain phenotypic fidelity, and ensure experimental and therapeutic reproducibility across diverse cell systems.
Scientific Principles: Dehydration and Ice Formation Dynamics
Fundamental Cryobiology of Freezing
When cells undergo freezing, they encounter a series of physical challenges that dictate their survival. The process begins with extracellular ice formation, which increases the solute concentration in the unfrozen fraction, creating an osmotic gradient that drives water efflux from the cell interior. This cellular dehydration is a protective mechanism that reduces the potential for intracellular ice formation (IIF), which is almost universally lethal to cells [4]. The relationship between cooling rate and cell survival follows a classic inverted U-shape curve, where excessively slow cooling causes prolonged exposure to hypertonic solutions and "solution effects" injury, while excessively rapid cooling results in damaging intracellular ice. The optimal cooling rate achieves a balance between these competing damaging mechanisms.
The cell membrane serves as the primary barrier regulating water transport during freezing. The kinetics of water movement across this membrane are governed by the membrane's hydraulic conductivity and the activation energy for water flow, both of which are temperature-dependent parameters. Mathematical models describing water transport have become increasingly sophisticated, enabling researchers to predict optimal cooling rates for specific cell types based on their biophysical characteristics. For most mammalian cells, including many sensitive cell types, the optimal cooling rate falls between 0.3°C and 3°C per minute, though specific cell types may require protocol individualization [41] [42].
Advanced Research: Controlled Ice Nucleation
Recent research has illuminated the critical role of ice nucleation temperature in determining freezing outcomes. A 2024 study investigating T cell cryopreservation demonstrated that controlling the ice nucleation temperature significantly influences intracellular dehydration patterns and subsequent viability. Using a model T cell line (Jurkat cells) in commercially relevant cryoformulations, researchers compared controlled nucleation at -6°C versus -10°C and uncontrolled spontaneous nucleation [27] [4].
The findings revealed that initiating ice formation at a higher temperature (-6°C), closer to the equilibrium freezing point of the cryoformulation, resulted in more extensive intracellular dehydration but substantially less intracellular ice formation during subsequent cooling. This controlled approach produced more consistent temperature profiles across product batches and improved post-thaw membrane integrity and viability compared to both lower nucleation temperatures and uncontrolled nucleation. The correlation between enhanced dehydration, reduced IIF, and improved cell survival underscores the importance of managing the initial freezing event, not just the subsequent cooling rate [27].
Standard Protocols for Sensitive Cell Types
Universal Pre-Freezing Preparation
Proper preparation before initiating controlled-rate freezing significantly influences post-thaw outcomes. The following pre-freezing requirements apply broadly across sensitive cell types:
Cell Health and Density: Cells should exhibit >90% viability before freezing and be harvested during their logarithmic growth phase at 70-80% confluence. This ensures metabolic competence and adequate energy reserves for cryopreservation stress. Target a freezing density of approximately 1-10 million cells/mL, adjusting for specific cell type requirements [41] [42].
Cryoprotectant Selection and Equilibration: Dimethyl sulfoxide (DMSO) remains the gold standard cryoprotectant for most sensitive cell types at concentrations typically ranging from 5% to 10% (v/v). Gradually add pre-cooled cryoprotectant solution to cell pellets to minimize osmotic shock. Allow equilibration for 10-15 minutes at 4°C to enable adequate permeation while limiting toxic exposure [41].
Container and Temperature Management: Use cryovials certified for ultra-low temperature storage. Maintain samples on ice during preparation and loading into the controlled-rate freezer to prevent premature warming or inconsistent starting conditions [41].
Cell-Type Specific Protocols
T Cells and CAR-T Cells
T cells used in immunotherapies represent a particularly sensitive cell population with specific protocol requirements. The following optimized protocol is adapted from recent research on Jurkat cells and primary T cells:
Table 1: Standard Controlled-Rate Freezing Protocol for T Cells
| Parameter | Specification | Rationale |
|---|---|---|
| Cryoprotectant | 5-10% DMSO in Plasma-Lyte A or equivalent | Balanced cryoprotection with clinical compatibility |
| Cooling Rate 1 | 1°C/min to -6°C | Slow cooling to nucleation temperature |
| Ice Nucleation | Controlled initiation at -6°C | Promotes dehydration, minimizes intracellular ice |
| Hold Time | 5-10 minutes at -6°C | Allows for cellular dehydration |
| Cooling Rate 2 | 1°C/min to -40°C to -50°C | Continued controlled cooling |
| Cooling Rate 3 | 10°C/min to -90°C | Rapid transition through dangerous temperature zone |
| Transfer Temp | Immediately to -150°C or liquid nitrogen | Prevents ice recrystallization |
This protocol leverages the benefits of controlled ice nucleation, which research has demonstrated enhances intracellular dehydration while minimizing intracellular ice formation in T cells. The hold time following nucleation allows for adequate water efflux before further temperature reduction [27] [4].
Mesenchymal Stem Cells (MSCs)
Mesenchymal stem cells require special consideration due to their differentiation potential and sensitivity to cryoprotectant toxicity. Research indicates that combining DMSO with hydroxyethyl starch (HES) permits reduction of DMSO concentration while maintaining viability:
Table 2: Standard Controlled-Rate Freezing Protocol for MSCs
| Parameter | Specification | Rationale |
|---|---|---|
| Cryoprotectant | 5% DMSO + 5% HES | Reduced DMSO toxicity with macromolecular support |
| Cooling Rate | 1°C/min to -80°C | Standard slow cooling for stem cells |
| Alternative Method | "Straight freeze" at -80°C | Simpler approach with comparable results for some lines |
| HES Molecular Weight | 109-609 kDa | Minimal impact of molecular weight on efficacy |
| Assessment Timing | 24+ hours post-thaw | Delayed assessment more accurately reflects recovery |
Notably, studies on rat MSCs have demonstrated that cryopreservation effects should be assessed at least 24 hours post-thaw rather than immediately after thawing, as this delayed assessment more accurately reflects true recovery and function. Additionally, the molecular weight of HES appears to play only a minor role in its cryoprotective efficacy [43].
Research across multiple cell types provides quantitative insights into optimal freezing parameters and their impact on viability outcomes:
Table 3: Comparative Freezing Parameters and Outcomes Across Cell Types
| Cell Type | Optimal Cooling Rate | Cryoprotectant Composition | Post-Thaw Viability | Key Findings |
|---|---|---|---|---|
| T Cells (Jurkat) | 1°C/min to -6°C, then 1°C/min to -50°C, 10°C/min to -90°C | 2.5-5% DMSO in Plasma-Lyte A | Significantly improved with controlled nucleation at -6°C | Higher nucleation temperature (-6°C) promoted dehydration, reduced intracellular ice [27] |
| Mesenchymal Stem Cells | 1°C/min to -80°C or "straight freeze" | 5% DMSO + 5% HES | Maintained with reduced DMSO | HES enables DMSO reduction without compromising viability [43] |
| General Mammalian Cells | -1°C to -3°C per minute | 10% DMSO standard | >90% with optimization | Cooling rate critical to balance dehydration and ice formation [42] |
Essential Materials and Reagents
Successful implementation of controlled-rate freezing protocols requires specific quality-controlled materials and equipment:
Table 4: Essential Research Reagent Solutions for Controlled-Rate Freezing
| Reagent/Equipment | Function | Application Notes |
|---|---|---|
| DMSO (Cell Culture Grade) | Penetrating cryoprotectant that disrupts hydrogen bonding to prevent ice crystal formation | Use high-purity, sterile-filtered grade; concentration typically 5-10% depending on cell type [42] [24] |
| Hydroxyethyl Starch (HES) | Non-penetrating cryoprotectant that modulates extracellular ice formation and provides osmotic buffering | Enables reduction of DMSO concentration; molecular weight (109-609 kDa) shows minimal impact on efficacy [43] |
| Controlled-Rate Freezer | Programmable cooling device that maintains specified temperature descent rates | Essential for reproducible cooling profiles; liquid nitrogen-free models available [4] |
| Cryovials (Certified) | Secure containment system for ultra-low temperature storage | Use vials certified for liquid nitrogen storage; check for microcracks before use [41] |
| Plasma-Lyte A or Similar | Balanced salt solution base for cryoprotectant formulations | Provides physiological ion balance; superior to simple saline for sensitive cells [27] |
Methodological Workflow
The following diagram illustrates the complete controlled-rate freezing workflow, highlighting the critical decision points and procedures that ensure optimal cell viability:
Technological Innovations and Future Perspectives
The field of controlled-rate freezing continues to evolve with several emerging technologies poised to enhance protocol standardization and outcomes for sensitive cell types. Automated freezing systems with AI-driven cooling algorithms represent a significant advancement, enabling real-time adjustment of freezing parameters based on cell type-specific requirements and potentially improving post-thaw viability [24]. These systems leverage machine learning analysis of historical viability data to optimize cryoprotectant formulations and cooling profiles.
Ice-free vitrification techniques offer an alternative approach that completely avoids ice crystal formation by ultra-rapid cooling to a glass-like state. While particularly valuable for especially sensitive cells like oocytes and embryos, this method presents challenges in scaling for larger volume samples. Similarly, nanoparticle-based cryoprotectants are under investigation as potential alternatives to reduce or replace DMSO, addressing concerns about its cellular toxicity and clinical side effects [24].
The integration of controlled ice nucleation methodologies into standard protocols represents another significant innovation, with research consistently demonstrating its benefits for T cells and other sensitive populations. As these technologies mature, they will likely become incorporated into Good Manufacturing Practice (GMP) standards for cell therapy products, enhancing batch-to-batch consistency and overall product quality in clinical applications [4].
Cryopreservation serves as a fundamental process for maintaining the viability and functionality of cellular therapeutics and active pharmaceutical ingredients during storage and transport. As a living medicine, these advanced therapy medicinal products (ATMPs) require sophisticated preservation techniques that conventional pharmaceuticals do not. The process of ice formation during freezing presents a critical challenge: uncontrolled ice nucleation leads to extensive supercooling, resulting in variable ice crystal structures that compromise product quality and batch consistency. Supercooling occurs when a liquid remains in a metastable state below its equilibrium freezing point without solidifying, creating conditions for potentially damaging ice formation.
Controlled ice nucleation has emerged as a powerful technological approach to manage the phase transition of water from liquid to solid. By artificially initiating ice formation at a predetermined temperature, this method significantly reduces the stochastic nature of supercooling, leading to more uniform ice crystal morphology and improved batch homogeneity. The implications for pharmaceutical development and cell therapy manufacturing are substantial, ranging from enhanced post-thaw cell viability to more consistent product characteristics across manufacturing batches. Within the broader context of freezing rate research, controlled nucleation represents a paradigm shift in understanding how to manage intracellular dehydration and ice formation—two critical factors that determine the success of cryopreservation protocols.
The Science of Supercooling and Ice Nucleation
Fundamental Principles
Ice nucleation encompasses the initial molecular events that trigger the phase transition from liquid water to solid ice. This process begins when water molecules collide, aggregate, and form clusters that eventually reach a critical size capable of stabilizing and promoting further ice crystal growth. Nucleation occurs only after the temperature drops below the solution's equilibrium freezing point, generating varying degrees of supercooling in the remaining liquid portion of the sample [44]. The latent heat of fusion—energy released during the phase change from water to ice—causes transient warming of the sample, a phenomenon that must be carefully managed in controlled freezing protocols [44].
Two distinct nucleation pathways exist in cryopreservation:
Homogeneous nucleation occurs spontaneously in the absence of any nucleating agents when water molecules arrange into ice crystals purely through statistical fluctuations, typically at temperatures below -38°C for pure water [45]. This process produces numerous small ice crystals but requires extensive supercooling, which may be detrimental to cellular systems.
Heterogeneous nucleation takes place when ice crystals form on the surface of ice-nucleating particles (INPs) or specific substrates, occurring at higher temperatures (-3°C to -10°C) with less supercooling [46]. This mechanism generates larger, more structured ice crystals with less intracellular ice formation.
The temperature difference between the actual freezing point and the equilibrium freezing point defines the degree of supercooling, which directly influences ice crystal size, distribution, and ultimately, cell viability post-thaw [47].
Impact on Cellular Systems
During freezing, ice formation initiates extracellularly, creating a concentrated solute environment that draws water out of cells through osmosis. The rate of cooling determines whether this water transport occurs efficiently or results in intracellular ice formation (IIF). Slow cooling permits sufficient time for cellular dehydration, while rapid cooling traps water inside cells, leading to lethal IIF [48]. The damaging effects extend beyond immediate cell death; even sublethal ice crystal damage can compromise therapeutic functions through mechanisms such as:
- Mechanical injury from extracellular ice crystals
- Osmotic stress from concentrated solutes
- Ionic imbalance disrupting cellular homeostasis
- Membrane disruption from ice crystal penetration
Recent research on hematopoietic stem and progenitor cells (HSPCs), effector T cells, and mesenchymal stromal/stem cells (MSCs) demonstrates that cryopreservation impacts different cell types variably, with some requiring specific freezing and thawing protocols to maintain functionality [48].
Experimental Evidence and Quantitative Data
T Cell Cryopreservation Studies
A 2024 investigation into T cell cryopreservation provides compelling quantitative evidence supporting controlled ice nucleation. The study systematically evaluated Jurkat cells (a model T cell line) in commercially relevant cryoformulations containing 2.5% and 5% v/v DMSO in Plasma-Lyte A [27]. Using a cryomicroscopic setup to monitor dynamic changes during freeze-thaw cycles, researchers correlated intracellular events with post-thaw viability outcomes.
Table 1: Impact of Ice Nucleation Temperature on T Cell Freeze-Thaw Parameters
| Nucleation Temperature | Intracellular Dehydration | Intracellular Ice Formation | Membrane Integrity | Post-Thaw Viability |
|---|---|---|---|---|
| -6°C (High) | More extensive | Less extensive | Highest preservation | Optimal recovery |
| -10°C (Lower) | Moderate | Moderate | Moderate preservation | Reduced recovery |
| Uncontrolled nucleation | Variable | More extensive | Most compromised | Lowest and most variable |
The cryomicroscopic studies revealed that an ice nucleation temperature of -6°C, close to the equilibrium freezing temperatures of the cryoformulations, led to more intracellular dehydration and less intracellular ice formation during freezing compared to either a lower ice nucleation temperature (-10°C) or uncontrolled ice nucleation [27]. The consistency of these findings across both bulk cryopreservation and single-cell observations underscores the robustness of controlled nucleation approaches.
Pharmaceutical Freeze-Drying Applications
Beyond cell therapy, controlled nucleation demonstrates significant value in pharmaceutical freeze-drying processes. A 2019 study compared ice fog methods and monitored controlled nucleation success after freeze-drying, with particular focus on how the ice nucleation temperature (TN) influences final product characteristics [49].
Table 2: Impact of Nucleation Temperature on Lyophilized Product Properties
| Nucleation Parameter | Specific Surface Area | Residual Moisture Content | Batch Homogeneity | Non-Nucleated Vials Risk |
|---|---|---|---|---|
| TN = -3°C | Moderate | Lower | Higher | Increased risk |
| TN = -10°C | Variable | Higher | Lower | Reduced risk |
The research concluded that TN is not the only specific surface area determining factor and that a high TN does not necessarily lead to larger pores but poses a higher risk of not-nucleating vials [49]. This highlights the delicate balance required in optimizing nucleation parameters for specific applications.
Methodologies and Experimental Protocols
Controlled Nucleation for T Cell Cryopreservation
The following detailed methodology outlines the protocol used in the 2024 T cell cryopreservation study [27], providing a reproducible framework for implementing controlled ice nucleation:
Materials Preparation:
- Prepare cryoformulations of 2.5% and 5% v/v DMSO in Plasma-Lyte A
- Culture Jurkat T cells to log-phase growth (approximately 1×10^6 cells/mL)
- Harvest cells by centrifugation at 300 × g for 5 minutes
- Resuspend cell pellet in cryoformulation to achieve 5-10×10^6 cells/mL
- Aliquot 1 mL cell suspensions into cryovials
Controlled Freezing Protocol:
- Place cryovials in a controlled rate freezer (CRF) pre-cooled to 4°C
- Cool samples at -1°C/minute to the target nucleation temperature
- For controlled nucleation at -6°C, hold samples for 2-5 minutes
- Implement nucleation trigger using appropriate method (ice fog or mechanical)
- After nucleation confirmation, continue cooling at -1°C/minute to -40°C
- Increase cooling rate to -5°C/minute to final temperature of -80°C
- Transfer samples to liquid nitrogen storage (-196°C) or vapor phase storage (-150°C to -170°C)
Thawing and Assessment:
- Rapidly thaw cells in a 37°C water bath with gentle agitation
- Dilute cell suspension drop-wise with pre-warmed culture medium
- Assess membrane integrity via trypan blue exclusion
- Evaluate post-thaw viability using flow cytometry with Annexin V/PI staining
- Measure functional recovery through cell proliferation assays
Freeze-Casting for Material Science Applications
Though derived from material science, the freeze-casting methodology offers valuable insights into controlling ice nucleation for structural applications [50]:
Perforated Substrate Method:
- Modify substrate surfaces with precise perforations to create nucleation sites
- Prepare aqueous slurries (e.g., graphene oxide dispersions) for freeze-casting
- Apply slurry to the perforated substrate in a controlled environment
- Implement directional freezing with defined temperature gradients
- Monitor ice crystal growth using microscopic imaging
This approach demonstrated remarkable efficacy, reducing the temperature range of ice nucleation from 18.8°C to 5.2°C when freeze-casting graphene oxide dispersions compared to pristine control substrates [50]. The significant reduction in nucleation variability directly translated to more consistent material structures with fewer disordered transitional zones.
Visualization of Processes and Workflows
Comparative Freezing Outcomes Diagram
Experimental Workflow Diagram
Research Reagents and Essential Materials
Successful implementation of controlled ice nucleation requires specific reagents and equipment designed to manage the freezing process with precision. The following table details key research solutions and their functions in cryopreservation protocols:
Table 3: Essential Research Reagents and Equipment for Controlled Nucleation Studies
| Category | Specific Product/Model | Function | Application Context |
|---|---|---|---|
| Cryoprotectants | Dimethyl sulfoxide (DMSO) | Permeating CPA preventing intracellular ice formation | T cell cryopreservation [27] [44] |
| Cryoprotectants | Glycerol | Permeating CPA with reduced toxicity | Sensitive cell types [44] |
| Cryoprotectants | Sugars (sucrose, trehalose) | Non-permeating CPA providing extracellular protection | Stabilization during freezing [44] |
| Cryoprotectants | Ice-binding proteins (IBPs) | Modify ice crystal structure and growth | Alternative to chemical CPAs [44] |
| Base Media | Plasma-Lyte A | Isotonic solution maintaining physiological pH | Cryoformulation base [27] |
| Equipment | Controlled-rate freezer (e.g., CryoMed CRF) | Programmable freezing with precise rate control | Reproducible nucleation protocols [47] |
| Equipment | Cryomicroscopy systems | Real-time visualization of ice formation | Protocol optimization [27] |
| Nucleation Inducers | Ice fog generators | Provide nucleation sites at defined temperatures | Pharmaceutical freeze-drying [49] |
| Nucleation Inducers | Perforated substrates | Engineered surfaces promoting nucleation | Freeze-casting applications [50] |
| Assessment Tools | Flow cytometer with Annexin V/PI | Quantitative viability and apoptosis measurement | Post-thaw recovery assessment [27] |
Implications for Cellular Therapeutics and Pharmaceutical Development
The strategic implementation of controlled ice nucleation carries profound implications for the development and commercialization of cellular therapeutics and pharmaceutical products. For Advanced Therapy Medicinal Products (ATMPs), including hematopoietic stem cells, mesenchymal stromal cells, effector T cells, and chimeric antigen receptor (CAR)-modified cell products, consistency in cryopreservation outcomes translates directly to predictable therapeutic efficacy and enhanced patient safety [48].
Current challenges in cell therapy manufacturing include the variable engraftment capacity of freeze-thawed cells and inconsistent functionality between production batches. Research indicates that certain cell types, particularly MSCs, may operate through a "hit and run" mechanism without requiring long-term engraftment, yet their therapeutic potency remains susceptible to cryopreservation-induced damage [48]. Controlled nucleation addresses this vulnerability by minimizing ice crystal injury to cell membranes and intracellular structures, thereby preserving critical biological functions.
From a commercial perspective, the batch homogeneity achieved through controlled nucleation supports regulatory compliance and quality assurance mandates for pharmaceutical manufacturing. The reduced variability between product batches decreases validation burdens and enhances manufacturing efficiency, ultimately contributing to more economically viable cellular medicines [48] [49]. Furthermore, the extended stability profiles enabled by optimized cryopreservation protocols facilitate distributed manufacturing models and global supply chains for cell-based therapies, expanding patient access to these innovative treatments.
As cryopreservation science continues to evolve, integration of controlled nucleation with advanced cryoprotectant formulations and precision thawing methodologies will likely establish new standards for preserving cellular function. The ongoing refinement of these techniques promises to accelerate the translation of cellular therapeutics from preclinical proof-of-concept to clinically effective and commercially sustainable medicines.
Ice nucleation, the initial process of ice crystal formation from a supercooled liquid, is a critical phenomenon with profound implications across numerous scientific and industrial fields. This technical guide focuses on three primary methods for inducing controlled ice nucleation: ice seeding, pressure shift, and the use of chemical nucleants. The control of this phase transition is not merely an academic exercise; it is a crucial determinant in the outcomes of processes ranging from the lyophilization of pharmaceuticals to the cryopreservation of biologics [51]. Within the specific context of cell therapy and biologics preservation, the rate of freezing, directly governed by the nucleation temperature, is a key variable in a broader thesis investigating its effect on cell dehydration and intracellular ice formation [27]. Uncontrolled, stochastic nucleation can lead to inconsistent product quality, while mastered nucleation protocols ensure batch-to-batch reproducibility, enhanced product stability, and ultimately, the safety and efficacy of sensitive biological products [51] [27].
Core Principles of Ice Nucleation
At its core, heterogeneous ice nucleation requires the presence of ice-nucleating particles (INPs) or specific techniques to overcome the energy barrier to crystal formation. The temperature at which nucleation occurs is a primary factor influencing ice crystal size and morphology. Higher nucleation temperatures (closer to 0°C) generally result in fewer, larger ice crystals, while lower nucleation temperatures produce a more numerous, finer ice crystal structure [51]. This fundamental principle directly impacts the freezing rate dynamics experienced by cells and biomolecules. During freezing, cells undergo dehydration as water is sequestered into extracellular ice; the rate of this process determines whether the cell experiences a protective, gradual dehydration or a damaging intracellular ice formation [27]. Controlled nucleation protocols are therefore designed to dictate this critical initial step, steering the process toward the desired outcome.
Practical Methods for Inducing Ice Nucleation
Pressure Shift Nucleation
The pressure shift nucleation method leverages the thermodynamic principle that the equilibrium freezing point of water decreases under elevated pressure. This technique is notably implemented in commercial systems like the ControLyo technology for pharmaceutical lyophilization [51].
- Basic Principle: A product is cooled under pressure to a temperature where it remains liquid, despite being below its atmospheric pressure freezing point. A rapid depressurization then creates a significant, uniform supercooling across the entire product volume, triggering instantaneous and homogeneous nucleation [51].
- Experimental Protocol: The following steps outline a standard protocol for implementing pressure shift nucleation in a lyophilization cycle:
- Cooling Phase: Load the product vials on the lyophilizer shelf and cool them to a target temperature several degrees below the product's equilibrium freezing point, but still above its pressure-depressed freezing point. A typical target is between -5°C and -10°C [51].
- Pressurization Phase: Introduce an inert gas (e.g., nitrogen or argon) into the lyophilization chamber to elevate the pressure to a predefined level, often in the range of 15-30 psig.
- Equilibration Phase: Hold the product under pressure for a brief period (e.g., 5-15 minutes) to ensure thermal equilibrium throughout all vials.
- Depressurization Phase: Rapidly release the chamber pressure to atmospheric levels. This sudden pressure drop induces a high degree of supercooling, resulting in simultaneous ice nucleation across the entire batch.
- Freezing Completion: Following nucleation, continue cooling the shelf to the final desired product freeze temperature (e.g., -40°C to -50°C) to complete the solidification process.
- Key Advantages:
- Batch Uniformity: Eliminates the random, stochastic nature of spontaneous nucleation, ensuring identical freezing conditions for every vial [51].
- Process Control: Enables a Quality by Design (QbD) approach for the freezing step, a critical process parameter in lyophilization [51].
- Improved Product Characteristics: Typically results in larger, more uniform ice crystals, which creates a product with higher porosity, reduced mass transfer resistance, and consequently, shorter primary drying times [51].
Chemical Nucleants
Chemical nucleants are substances that provide a template for ice crystal formation, thereby reducing the energy barrier to nucleation. A prominent category includes specific ice-nucleating bacteria, such as certain strains of Pseudomonas syringae [52].
- Basic Principle: These organisms produce a membrane-bound protein that structurally mimics an ice crystal lattice, facilitating the orderly arrangement of water molecules into ice at relatively high temperatures (up to -2°C) [52].
- Experimental Protocol:
- Nucleant Preparation: Obtain a purified preparation of the ice-nucleating protein or a suspension of the nucleating bacteria. The material may require dilution in an appropriate buffer to achieve the desired nucleation activity.
- Introduction to Sample: Introduce a small, controlled volume of the nucleant preparation into the sample solution. This can be done by direct pipetting or by pre-treating surfaces.
- Incubation and Cooling: Cool the sample at a controlled rate. Nucleation will occur at a characteristic temperature dictated by the efficiency of the nucleant used.
- Key Advantages and Applications:
- Considerations: The use of biological material, particularly viable pathogens, is inappropriate for pharmaceutical products or cell therapy cryopreservation due to contamination risks.
Ice Seeding
Ice seeding is a classic laboratory technique where a microscopic ice crystal is manually introduced into a supercooled sample to initiate crystallization.
- Basic Principle: By directly adding a pre-formed ice crystal, the energy barrier for nucleation is bypassed entirely, allowing controlled crystal growth to begin immediately at a defined temperature.
- Experimental Protocol:
- Supercooling Phase: Cool the sample to a specific, target supercooled temperature (e.g., -2°C to -5°C for many aqueous solutions). Maintain temperature stability.
- Seed Introduction: Using a pre-chilled tool (e.g., a spatula, wire loop, or pipette tip containing a small ice crystal), quickly touch the surface of the supercooled liquid or inject a minute volume of ice-slurry.
- Nucleation Confirmation: Visual confirmation of instantaneous crystal propagation throughout the sample verifies successful seeding.
- Key Advantages:
- Simplicity and Low Cost: Requires no specialized equipment.
- Precise Timing: Allows the researcher to dictate the exact moment of nucleation.
- Challenges:
- Lack of Scalability: Difficult to implement uniformly in large-scale or high-throughput industrial processes.
- Operator Dependency: Success and reproducibility are highly dependent on operator skill, introducing a potential source of variability.
- Contamination Risk: The manual introduction of a seed carries a risk of contaminating the sample.
Comparison of Nucleation Methods
Table 1: Comparative analysis of key ice nucleation induction methods.
| Method | Principle | Typical Nucleation Temperature | Scalability | Primary Applications | Key Advantages |
|---|---|---|---|---|---|
| Pressure Shift | Thermodynamic depression of freezing point via pressure. | Programmable, often -5°C to -10°C [51] | Excellent for commercial-scale batch processing. | Pharmaceutical Lyophilization [51] | Superior batch uniformity; amenable to QbD; shorter drying times. |
| Chemical Nucleants | Biological/protein template mimics ice lattice. | High, up to -2°C [52] | Moderate to High (depending on nucleant) | Snow-making, Food Texturing, Atmospheric Research [52] | Very high nucleation temperature; effective for specific applications. |
| Ice Seeding | Manual introduction of an ice crystal. | Determined by user (e.g., -2° to -5°C) | Low (laboratory-scale only) | Basic Research, Small-scale Lab Protocols | Simple and inexpensive; precise timing. |
Impact on Freezing Rates, Cell Dehydration, and Ice Formation
The choice of nucleation method directly influences the subsequent freezing trajectory, with critical consequences for cellular systems. Research has quantitatively demonstrated the link between nucleation temperature and cell viability.
A 2024 study on T-cell cryopreservation provides a compelling experimental model within the user's thesis context. The study compared controlled ice nucleation at -6°C against both uncontrolled nucleation and nucleation at -10°C. The findings were significant: the higher nucleation temperature of -6°C, which is closer to the solution's equilibrium freezing temperature, resulted in markedly increased intracellular dehydration and a corresponding reduction in intracellular ice formation during the freezing process. This controlled cellular response directly translated to improved biological outcomes: bulk cryopreservation experiments confirmed that this protocol yielded superior cell membrane integrity and post-thaw viability [27]. This evidence strongly supports the thesis that controlling the ice nucleation temperature is a powerful lever to manage the trade-off between dehydration and ice formation, ultimately dictating the success of cryopreservation protocols for sensitive biologics like cell therapies.
The Scientist's Toolkit
Table 2: Essential research reagents and materials for ice nucleation studies.
| Item | Function/Application |
|---|---|
| Controlled Rate Freezer | A programmable instrument that enforces a user-defined cooling profile, essential for standardizing freezing protocols in cryopreservation and lyophilization development [27]. |
| Cryomicroscopy Setup | Allows for the direct, real-time observation of dynamic cellular events during freezing and thawing, such as intracellular dehydration and ice formation [27]. |
| Differential Scanning Calorimetry (DSC) | Used to determine key thermodynamic parameters of cryoformulations, including the equilibrium freezing temperature and glass transition temperature, which are critical for protocol design [27]. |
| Portable Ice Nucleation Chamber (e.g., MRINC) | An advanced research tool that integrates a climate chamber with holographic microscopy for real-time detection and differentiation of nano-sized ice crystals and supercooled droplets in atmospheric studies [53]. |
| Cryoprotective Agents (e.g., DMSO) | Compounds added to solutions to protect cells from freeze-related damage by reducing ice crystal growth and mitigating osmotic shock. Typical concentrations range from 2.5% to 10% v/v [27]. |
Experimental Workflow and Data Analysis
The following diagram illustrates a generalized experimental workflow for studying the impact of controlled ice nucleation in cell cryopreservation, integrating key tools and measurements.
For data interpretation, particularly from drop-freezing experiments, the HUB (heterogeneous underlying-based) method provides a robust framework. This computational approach models the underlying distribution of ice nucleation temperatures (Pu(T)) from experimental fraction frozen (fice(T)) data. The HUB-forward code predicts freezing curves from a hypothesized Pu(T), while the HUB-backward code extracts the analytical differential freezing spectrum nm(T) from experimental data, enabling the identification and characterization of IN subpopulations in complex biological samples [54].
Cryopreservation is a cornerstone of modern biotechnology and medicine, enabling the long-term storage of biological specimens for applications ranging from basic research to clinical therapies. The fundamental challenge lies in navigating the physical processes of ice formation and dehydration during freezing and thawing. As described by the two-factor hypothesis of freezing injury, cell survival depends on a delicate balance: cooling must be slow enough to avoid lethal intracellular ice formation, yet fast enough to prevent excessive cell dehydration due to osmotic stress [55] [56]. While this principle provides a general framework, optimal cryopreservation parameters vary significantly between cell types due to their distinct biological and physical characteristics. T-cells, induced pluripotent stem cells (iPSCs), and other biologics each possess unique membrane permeability, size, function, and sensitivity to cryoprotectant agents (CPAs), necessitating tailored approaches. This technical guide examines cell-type-specific considerations, providing detailed methodologies and quantitative data to optimize cryopreservation protocols within the broader context of freezing rate effects on cell dehydration and ice formation.
Theoretical Foundation: Modeling Cell-Scale Freezing Processes
Advanced mathematical models provide the theoretical framework for understanding intracellular dynamics during cryopreservation. These models simulate the coupled transport of water and CPAs across cell membranes, as well as the nucleation and growth of intracellular ice crystals, offering predictive insights for protocol optimization.
Evolution of Cryopreservation Models
The foundational model developed by Mazur et al. simulated the impact of cooling rates on cellular dehydration and intracellular ice formation, establishing the fundamental two-factor hypothesis [55]. Toner et al. later proposed a physicochemical model based on modified classical heterogeneous nucleation theory to analyze intracellular ice nucleation during freezing, considering both surface-catalyzed and volume-catalyzed nucleation [55]. Subsequent improvements by Karlsson et al. incorporated a non-isothermal, diffusion-limited crystal growth model accounting for the variation in intracellular solution concentration with temperature [55]. Zhao et al. further refined this approach by considering the influence of intracellular ice volume on subsequent water transport and ice growth [55].
More recent models have increased in sophistication. Weng et al. developed non-isothermal equations to investigate the coupled transport of water and permeable CPA in non-ideal and non-dilute solutions [55]. The Surface Area Regulation (SAR) model proposed by Traversari et al. couples osmotic behavior with cellular mechanics and the regulation of cell membrane surface area, providing a more accurate description of cell osmotic behavior [55]. The most comprehensive models, such as that presented by Yuan et al., now incorporate descriptions of recrystallization during the rewarming period, making the description of intracellular crystallization phenomena more consistent with physical reality [55].
Table 1: Key Cryopreservation Models and Their Features
| Model | Key Features | Limitations |
|---|---|---|
| Mazur Model | Establishes foundation of two-factor hypothesis; links cooling rates to dehydration and intracellular ice | Neglects CPA permeation and intracellular crystallization [55] |
| Toner Model | Applies heterogeneous nucleation theory; analyzes surface and volume-catalyzed nucleation | Does not describe recrystallization during rewarming [55] |
| Karlsson/Zhao Model | Non-isothermal, diffusion-limited crystal growth; accounts for intracellular concentration changes | Lacks coupled transport of water and CPA [55] |
| Weng Model | Coupled transport in non-ideal, non-dilute solutions; more accurate transmembrane transport | No description of intracellular crystallization [55] |
| SAR Model | Couples osmotic behavior with cellular mechanics and membrane regulation | Limited description of crystallization processes [55] |
| Yuan et al. Model | Includes recrystallization during rewarming; comprehensive freeze-thaw simulation | Increased computational complexity [55] |
Visualizing the Freeze-Thaw Process and Cell Response
The following diagram illustrates the key physical processes and cellular responses during cryopreservation, integrating the principles from the theoretical models discussed.
The fundamental damage mechanisms during cryopreservation include ice crystallization pressure that can fracture surrounding structures [57], recrystallization during thawing that causes additional damage [55], and osmotic stress leading to cell dehydration or swelling [56]. The formation of ice crystals damages muscle cells and oxidizes components like protein and fat in biological tissues, resulting in quality deterioration and nutritional value loss [13]. During thawing, ice crystals melt into liquid water, but the muscle tissue has already been damaged by the ice crystals, preventing full recovery to the pre-frozen state [13].
Cell-Type-Specific Cryopreservation Protocols
T-Cells and Peripheral Blood Mononuclear Cells (PBMCs)
T-cells and PBMCs are critical components of immunotherapies such as CAR T-cell therapy. These cell types are typically cryopreserved using DMSO-based solutions, though research into DMSO-free alternatives is advancing.
Experimental Protocol for T-Cell Cryopreservation
Cell Preparation: Isolate PBMCs from leukocyte suspensions using Lymphocyte Separation Medium via centrifugation at 700 × g for 30 minutes with the brake off [58]. Collect the PBMC layer and wash with PBS three times at 500 × g for 5 minutes [58]. For T-cell subsets, stain with specific surface markers (CD3+ for T-cells, CD3+CD4+ for helper T-cells, CD3+CD8+ for cytotoxic T-cells) for identification and separation [59].
Freezing Solution Preparation: Prepare cryoprotectant solutions in Normosol-R. For DMSO-containing controls, use 10% DMSO solution. For DMSO-free formulations, optimize combinations of osmolytes such as Sucrose-Glycerol-Isoleucine (SGI), Trehalose-Glycerol-Isoleucine (TGI), or Maltose-Glycerol-Isoleucine (MGI) [59]. The concentration space of each osmolyte should be discretized to six levels with equal spacing, with the highest level set by either toxicity or solubility limits [59].
Freezing Procedure: Resuspend cells at 6 million cells/mL in the cryoprotectant solution [59]. Use a controlled rate freezer with the following cooling curve: (1) start at 20°C, (2) cool at -10°C/min to 0°C, (3) hold at 0°C for 15 minutes, (4) cool at -1°C/min to -8°C, (5) rapid cool at -50°C/min to -45°C, (6) warm at +15°C/min to -12°C, (7) cool at -1°C/min to -60°C, and (8) cool at -10°C/min to -100°C [59]. The rapid cooling and rewarming steps (5 and 6) are used to induce nucleation in the extracellular solution.
Storage and Thawing: Store frozen samples in vapor-phase liquid nitrogen for at least 24 hours. Thaw rapidly in a 37°C water bath until a small portion of ice remains [58]. Transfer the cell suspension to a tube containing pre-warmed RP10 medium (RPMI1640 with 10% FBS, 10 mM HEPES, and 0.1 mg/mL Gentamycin) [58]. Centrifuge at 500 × g for 5 minutes, resuspend in fresh medium, and assess viability.
Quantitative Recovery Data for T-Cells and PBMCs
Table 2: Post-Thaw Recovery of PBMC Subsets in Different Cryoprotectants
| Cell Type | Cryoprotectant | Post-Thaw Recovery | Viability Drop (24h post-thaw) | Key Observations |
|---|---|---|---|---|
| Helper T-Cells | DMSO-free (Optimized) | >80% | 17% | Significant difference in recovery between T-cell subsets [59] |
| Cytotoxic T-Cells | DMSO-free (Optimized) | >80% | 10% | Lower apoptosis rate compared to helper T-cells [59] |
| PBMCs (Overall) | 10% DMSO Control | ~70-80% | Not specified | Traditional benchmark for comparison [59] |
| PBMCs (scRNA-seq) | Optimized DMSO Protocol | Stable population | Minimal change | Maintained population composition after 6-12 months storage [58] |
Research indicates significant differences between post-thaw recovery for helper T-cells and cytotoxic T-cells, with statistical models showing that preferred concentration levels of osmolytes and interaction modes were distinct between these subsets [59]. Immediately post-thaw, both helper and cytotoxic T-cells had >30% of cells in early apoptosis, but after 24 hours the number of cells in early apoptosis decreased to below 20% [59].
Induced Pluripotent Stem Cells (iPSCs)
iPSCs present unique cryopreservation challenges due to their particular sensitivity to freezing and thawing processes. Their large surface area-to-volume ratio and membrane permeability make them highly vulnerable to intracellular ice formation.
Experimental Protocol for iPSC Cryopreservation
Cell Preparation: Culture iPSCs under standard conditions. Before freezing, confirm the absence of microbial contamination. To prevent Mycoplasma transfer, wear face masks during the procedure [56]. Passage cells as aggregates rather than single cells to maintain cell-cell contacts that support survival [56].
Freezing Solution Preparation: Use culture medium supplemented with 10% DMSO, which has an osmolarity of approximately 1.4 osm/L [56]. For enhanced stability, consider adding Ficoll 70 to the freezing solution, which enables storage at -80°C for at least one year without compromising viability and pluripotency [56].
Freezing Procedure: Implement a controlled freezing process with specific rates for different temperature zones. Hayashi et al. recommend a three-zone profile: (1) fast cooling in the dehydration zone, (2) slow cooling in the nucleation zone, and (3) fast cooling in the further cooling zone [56]. A rate of -1°C/min is frequently used for iPSCs and falls within the optimal range of -0.3 to -1.8°C/min identified for human embryonic stem cells [56]. Use cryocontainers for controlled-rate freezing in -80°C freezers before transfer to liquid nitrogen tanks or -150°C freezers for long-term storage.
Storage and Thawing: Store iPSCs in the vapor phase of liquid nitrogen (approximately -150°C to -160°C) or in -150°C freezers to prevent warming above the extracellular glass transition temperature of -123°C [56]. Thaw rapidly in a 37°C water bath. To prevent osmotic shock, carefully remove DMSO by gentle centrifugation and resuspension in fresh medium. Plate thawed cells on Matrigel-coated culture plates at appropriate density.
Critical Parameters for iPSC Cryopreservation
For optimal iPSC cryopreservation, several factors beyond basic protocol steps require attention:
- Cell Growth Phase: Freeze cells during the logarithmic growth phase for best recovery [56].
- Cooling Rate Control: Strictly control cooling rates as iPSCs are more vulnerable to intracellular ice formation than many other cell types [56].
- Temperature Monitoring: Avoid temperature fluctuations during storage, as warming above -123°C (extracellular glass transition temperature) or above -47°C (intracellular glass transition temperature) can stress cells and reduce viability [56].
- Post-Thaw Assessment: Allow 4-7 days for recovery post-thaw. If recovery problems occur, consider clone-to-clone variability and potential osmotic shock during thawing [56].
Biologics and Tissue Constructs
Biologics such as tissues and organs present additional complexities due to their three-dimensional structure. Vascularized composite tissues, corneas, ovaries, and even larger organs like hearts and kidneys have been targeted for cryopreservation to address clinical shortages [55]. The primary challenge lies in managing ice formation throughout complex tissue matrices without causing structural damage.
Experimental Protocol for Tissue Cryopreservation
Tissue Preparation: Prepare tissue samples of appropriate dimensions. For meat samples (as a model biological tissue), standardize size and shape to ensure consistent freezing profiles [13].
Freezing Methodology: Implement rapid freezing to produce small, uniform ice crystals. For direct freezing, ensure contact between the biological material and cryogenic liquids to significantly enhance thermal exchange efficiency [13]. Monitor temperature throughout the sample to ensure consistent freezing rates.
Storage and Thawing: Maintain consistent storage temperatures without fluctuations, as temperature variations lead to ice crystal growth and recrystallization, causing additional tissue damage [13]. For thawing, control the rate to minimize further damage. Note that during thawing, the temperature of muscle tissue is influenced by both ambient temperature and the water phase transition, which affects heat distribution and water status inside the muscle [13].
Assessment Methods: Use Low-Field Nuclear Magnetic Resonance (LF-NMR) to analyze water distribution and status in tissue matrices based on T2 relaxation times [13]. Employ microscopy to visualize ice crystal morphology and fractal dimension analysis to quantitatively analyze the irregular, complex shapes of ice crystals [13].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Cryopreservation Research
| Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Cryoprotectant Agents | DMSO, Glycerol, Propanediol, Methanol | Penetrate cells, reduce ice crystal formation | DMSO cytotoxicity varies by cell type; concentration optimization required [56] |
| Osmolytes (DMSO-free) | Sucrose, Trehalose, Maltose, Glycerol, Isoleucine | Stabilize biological systems, act as natural osmolytes | Optimal combinations cell-type specific; statistical modeling helpful [59] |
| Additives | Ficoll 70, Fetal Bovine Serum (FBS), HEPES | Enhance stability, provide nutrients, buffer pH | Ficoll 70 enables -80°C storage for iPSCs [56] |
| Culture Media | RPMI1640, Normosol-R | Maintain cell viability during processing | Composition affects osmotic balance [59] [58] |
| Separation Media | Lymphocyte Separation Medium | Isolate specific cell populations | Critical for PBMC preparation [58] |
| Viability Assays | Trypan Blue, Propidium Iodide, Calcein-AM, Annexin V | Assess cell viability, apoptosis | Multiple assays provide complementary data [59] [58] |
| Phenotyping Tools | Flow cytometry antibodies (CD3, CD4, CD8, CD45) | Identify specific cell types | Essential for heterogeneous populations [59] [58] |
The cryopreservation of diverse cell types requires both a fundamental understanding of freezing processes and specialized approaches tailored to specific cellular characteristics. T-cells benefit from optimized DMSO-free cryoprotectant combinations that recognize subset-specific responses, while iPSCs demand precise cooling rate control and specialized handling to preserve pluripotency. Biologics and tissues require management of ice crystallization throughout three-dimensional structures. In all cases, the careful balance between preventing intracellular ice formation and minimizing cellular dehydration remains paramount. As cryopreservation science advances, the development of more sophisticated models that incorporate recrystallization during rewarming and cellular mechanical properties will further enhance our ability to preserve the viability and function of diverse biological materials for research and clinical applications.
Solving the Freeze-Thaw Puzzle: Troubleshooting Low Viability and Process Optimization
The cryopreservation of cells is a cornerstone of modern biotechnology, enabling the advancement of cell therapies, regenerative medicine, and fundamental biological research. The success of these fields hinges on the post-thaw viability and functionality of preserved cells. However, a central challenge persists: cellular damage during the freeze-thaw cycle. This damage primarily manifests through two distinct mechanisms—intracellular ice formation (IIF) and solute damage (or solution effects injury)—which are governed by the cooling rate [60] [61]. Diagnosing the primary cause of failure is critical for refining cryopreservation protocols. A misinterpretation can lead researchers to adjust parameters in the wrong direction, further compromising cell survival. This guide provides researchers and drug development professionals with a detailed framework for differentiating between these two failure modes, supported by current experimental data and methodologies.
The core relationship between cooling rate and cellular injury was elegantly formalized by Mazur's "two-factor hypothesis" [61]. At low cooling rates, cells have sufficient time to lose water osmotically in response to extracellular ice formation. This prevents deadly intracellular ice but exposes cells to prolonged periods of high solute concentrations and extreme volume contraction, leading to solute damage. Conversely, at high cooling rates, there is insufficient time for water to leave the cell, resulting in severe supercooling and lethal intracellular ice formation [60] [62]. An optimal cooling rate exists that minimizes the combined injury from both mechanisms.
Theoretical Foundations of Freezing Damage
The Two Principal Mechanisms of Cryoinjury
Solute Damage (Solution Effects Injury) Solute damage occurs during slow cooling and is a consequence of the physical-chemical changes in the extracellular environment. As ice forms in the extracellular solution, dissolved solutes (e.g., salts, buffers) are excluded from the crystal lattice, becoming concentrated in the diminishing volume of unfrozen liquid [60]. This creates a hypertonic environment, driving water out of the cell and causing severe cellular dehydration and shrinkage. Injury is attributed to several factors:
- Elevated Electrolyte Concentrations: Denaturation of proteins and disruption of lipid bilayers due to the increased ionic strength [60].
- Cell Shrinkage: Excessive reduction in cell volume can cause irreversible damage to the plasma membrane and cytoskeleton [61].
- pH Changes: Alterations in the pH of the unfrozen fraction can disrupt enzyme activity and metabolic processes.
- Mechanical Stress: In tissues and dense cell suspensions, the growth of extracellular ice crystals can physically crush cells or disrupt tissue architecture [60].
Intracellular Ice Formation (IIF) IIF is almost invariably lethal and is associated with rapid cooling rates. When the cooling is too fast, the cell cannot dehydrate rapidly enough to maintain chemical potential equilibrium with the external environment. The intracellular solution becomes increasingly supercooled, creating a high thermodynamic driving force for ice nucleation. Ice can then form inside the cell, either through propagation (seeding) from the extracellular space or via homogeneous or heterogeneous nucleation [26] [62]. The damage from IIF is often mechanical; intracellular ice crystals can rupture organelles, the cytoskeleton, and the plasma membrane [60]. It has been suggested that the most severe damage may actually occur during thawing, through recrystallization processes [62].
The Role of Cell Type and Nucleation Temperature
The classic two-factor model is further complicated by cell-specific characteristics. Recent studies show that confluent cell monolayers can tolerate IIF better than single cells in suspension, suggesting that intracellular ice can, in some specific contexts, have a protective effect by preventing excessive dehydration during subsequent slow cooling [63]. This highlights that the cryobiological response is not universal.
A critical and often overlooked parameter is the extracellular ice nucleation temperature (Tnuc). Spontaneous nucleation leads to variable and often excessive supercooling, which increases the probability of IIF. Controlled ice nucleation, where ice formation is initiated at a specified temperature (e.g., -4°C to -6°C), has emerged as a key strategy to improve reproducibility and outcomes. By minimizing initial supercooling, controlled nucleation promotes more gradual cellular dehydration and significantly reduces the incidence of IIF [4] [27].
Table 1: Characteristic Features of Intracellular Ice vs. Solute Damage
| Feature | Intracellular Ice Formation (IIF) | Solute Damage (Solution Effects) |
|---|---|---|
| Primary Cause | Overly rapid cooling rate | Overly slow cooling rate |
| Key Event | Insufficient cellular dehydration; intracellular water freezes | Excessive cellular dehydration; prolonged exposure to concentrated solutes |
| Visual Cues (Cryomicroscopy) | Sudden darkening or "flashing" of the cell interior | Extreme and progressive cell shrinkage |
| Impact on Membrane | Mechanical rupture by ice crystals | Osmotic stress; phase transitions from liquid crystalline to gel state [62] |
| Impact on Proteins | Potential for mechanical shearing | Concentration-induced denaturation; relatively stable during freezing itself [62] |
| Post-Thaw Viability | Very low; often immediate lysis | Can be moderate but with reduced metabolic function |
Experimental Diagnostics and Methodologies
A multi-faceted approach is required to conclusively diagnose the mechanism of cryoinjury. The following experimental toolkit allows for direct observation, quantitative measurement, and molecular-level analysis.
Cryomicroscopy and Volume Analysis
Detailed Protocol:
- Cell Preparation: Suspend cells in the desired cryoprotectant solution (e.g., culture medium with 5-10% DMSO). For controlled experiments, use a model cell line like Jurkat (T-cells) or V-79W fibroblasts [63] [4].
- Sample Loading: Place a small volume (2-10 µL) of the cell suspension between two glass coverslips seated in a controlled environmental chamber on a microscope stage equipped with a programmable cooling unit.
- Freezing Protocol: Program the system to cool at a specific rate (e.g., 1°C/min to 10°C/min). For studies on nucleation temperature, use a controlled nucleation device (e.g., a pulse of pressurized gas or a cold finger) to initiate ice formation at a predetermined temperature (e.g., -5°C) [4] [27].
- Image Acquisition & Analysis: Use time-lapse phase contrast or polarized light microscopy to record cell behavior. Key metrics to track include:
- Time of Intracellular Ice Formation: A sudden change in optical contrast (darkening) indicates IIF [4].
- Cell Volume Change: Use image analysis software to measure the cross-sectional area of individual cells over time, calculating the volume reduction as a function of temperature.
This methodology directly visualizes the trade-off between dehydration and IIF. As demonstrated in a 2024 study on T-cells, a higher nucleation temperature (Tnuc = -6°C) led to significantly greater cellular dehydration but a lower incidence of IIF compared to a lower nucleation temperature (Tnuc = -10°C) [4] [27].
Biophysical Modeling of Water Transport and IIF
Computational models provide a quantitative framework to predict cell response. The classic water transport model is based on the work of Mazur and others [61]. The flux of water out of a cell, dV/dt, during freezing is given by:
dV/dt = Lp * A * RT * ln(Pₚ / Pₛ) / νw
Where:
- Lp is the membrane hydraulic conductivity
- A is the cell surface area
- R is the gas constant
- T is the temperature
- Pₚ and Pₛ are the vapor pressures of the intracellular and extracellular solutions, respectively
- νw is the molar volume of water
More advanced network models can simulate non-uniform dehydration and spatial ice growth patterns inside the cell, incorporating internal structures like the nucleus, which has different water permeability and diffusivity than the cytoplasm [26]. These models require cell-specific parameters for Lp and its activation energy (ELp), which can be determined experimentally.
Table 2: Quantitative Data from Key Cryopreservation Studies
| Cell Type / System | Cooling Rate / Nucleation Temp. | Key Measured Outcome | Reference |
|---|---|---|---|
| Jurkat (T-cells) | Tnuc = -6°C | ~40% more dehydrated, less IIF, higher viability | [4] [27] |
| Jurkat (T-cells) | Tnuc = -10°C | Less dehydrated, higher IIF, lower viability | [4] [27] |
| LNCaP (Prostate Tumor) | Tnuc = -6°C | Optimal survival; balances dehydration and IIF | [62] |
| LNCaP (Prostate Tumor) | Tnuc = -3°C | Co-operative membrane phase transition; lower conformational disorder | [62] |
| V-79W Hamster Fibroblasts (Single Cells) | Increased IIF incidence | Decreased post-thaw recovery | [63] |
| MDCK Cells (Confluent Monolayers) | Increased IIF incidence | Higher survival (protective effect of IIF) | [63] |
| Bovine Muscle | -80°C (Fast Freezing) | Small, even ice crystals; minor damage to muscle fibers | [64] |
| Bovine Muscle | -12°C (Slow Freezing) | Large, uneven ice crystals; severe structural damage | [64] |
Molecular-Level Analysis: FTIR Spectroscopy
Fourier Transform Infrared (FTIR) spectroscopy probes the molecular structure of cellular components during freezing, offering insights beyond viability assays.
Detailed Protocol:
- Sample Preparation: Concentrate cells into a pellet (~10 µL) and sandwich between two CaF₂ infrared windows with a thin spacer [62].
- Controlled Freezing: Mount the sample in a temperature-controlled stage/freezing holder. Cool at a controlled rate (e.g., 2°C/min) and use an ice nucleator (e.g., Pseudomonas syringae) to control the nucleation temperature.
- Spectral Acquisition: Collect spectra continuously during cooling and warming. Key regions to monitor include:
- Lipid Phase Behavior: The CH₂ symmetric stretching band (~2850 cm⁻¹). A shift to a higher wavenumber indicates a transition from the liquid crystalline to gel phase. The cooperativity of this transition is affected by dehydration and IIF [62].
- Protein Structure: The amide I (~1650 cm⁻¹) and amide III (~1200-1350 cm⁻¹) bands. Changes in secondary structure (α-helix to β-sheet) indicate denaturation, which is more closely linked to solute effects and heating than freezing itself [62].
This technique revealed that in LNCaP cells, nucleation at -3°C resulted in a more cooperative membrane phase transition than nucleation at -10°C, linking the physical state of the membrane directly to the freezing conditions [62].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagent Solutions for Cryoinjury Diagnostics
| Reagent / Material | Function & Application in Diagnosis |
|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating cryoprotectant; reduces IIF and solute concentration by forming hydrogen bonds with intracellular water [4]. A standard for comparison. |
| Plasma-Lyte A | Isotonic, physiological solution; used as a base for cryoformulations to avoid confounding effects from standard salt buffers [4]. |
| Pseudomonas syringae | A natural ice-nucleating agent; used to consistently induce controlled extracellular ice nucleation at high, defined temperatures (~-4°C to -8°C) [62]. |
| Acridine Orange / Propidium Iodide | Fluorescent viability stains for membrane integrity assays post-thaw; used to quantify viability in bulk freezing experiments [4]. |
| CaF₂ Infrared Windows | Material for FTIR spectroscopy; transparent to IR light and compatible with low-temperature sample holders [62]. |
| Programmable Controlled-Rate Freezer | Equipment that ensures a reproducible, user-defined cooling profile. Liquid nitrogen-free versions are now available for greater convenience [4]. |
Integrated Workflow for Diagnosis
The following diagram synthesizes the diagnostic pathway, integrating the experimental methods described above to conclusively identify the dominant mechanism of cryoinjury in a given scenario.
Diagnosing the root cause of cryopreservation failure is a systematic process that moves from observing macroscopic outcomes to probing microscopic and molecular events. The investigator must correlate cooling parameters with direct observational data from cryomicroscopy and support these findings with molecular-level insights from techniques like FTIR and predictive biophysical models. The emerging role of controlled ice nucleation as a critical parameter further refines our ability to steer cellular response away from both deleterious fates—towards dehydration without excessive solute damage, and away from the lethal formation of intracellular ice. By applying this integrated diagnostic framework, researchers can rationally develop robust, cell-specific freezing protocols that maximize survival and, crucially, maintain the functionality of these precious cellular resources.
Abstract The cryopreservation of complex cells, such as those used in advanced cell therapies, presents a significant bottleneck in the widespread application of regenerative medicine. The cooling rate profile is a critical process parameter that directly influences cell survival by governing the competing risks of intracellular ice formation and osmotic stress-induced dehydration. This whitepaper explores the application of a "Fast-Slow-Fast" cooling model, a multi-phasic approach designed to navigate these challenges for sensitive cell types. Within the broader thesis on the effect of freezing rates, this guide provides a detailed technical framework, including underlying biophysical principles, optimized experimental protocols, key reagents, and data analysis techniques to enable researchers to implement and validate this strategy for improving post-thaw viability and function.
The transition of cell therapies from the laboratory to the clinic is heavily dependent on robust cryopreservation protocols that maintain cell viability, potency, and functionality post-thaw. For complex cells—including induced pluripotent stem cells (iPSCs), their differentiated progeny (e.g., dopaminergic neurons, cardiomyocytes), and other primary cells—conventional slow-freeze protocols often yield suboptimal results [9] [7]. These cells are particularly vulnerable to the twin insults of cryopreservation: intracellular ice formation (IIF), which is lethal to cells, and excessive dehydration, which can cause osmotic shock and solute damage [44] [65].
The "Fast-Slow-Fast" model proposes a nuanced cooling profile that moves beyond a single, constant cooling rate. This approach aims to precisely manage the thermodynamic and biochemical phenomena during freezing by segmenting the process into distinct phases, each optimized for a specific protective goal. Its application is especially relevant within novel, off-the-shelf cell therapies, where the removal of cytotoxic cryoprotectants like dimethyl sulfoxide (Me₂SO) is not feasible prior to administration, thus placing greater emphasis on the physical freezing process itself to ensure cell health [9].
Theoretical Foundation: The Biophysics of Freezing
Successful cryopreservation hinges on controlling the phase change of water. As an aqueous solution cools below its equilibrium freezing point, it becomes supercooled before stochastic ice nucleation occurs. This is an exothermic process, releasing the latent heat of fusion [44].
- The Slow Freezing Paradigm and Its Limitations: The conventional slow-freeze protocol (approximately -1°C/min) is designed to promote extracellular ice formation, which concentrates the extracellular solutes. This creates an osmotic gradient that draws water out of the cell, thereby minimizing the chance of IIF. However, for many complex cells, this slow dehydration can itself be damaging, leading to excessive cell shrinkage, prolonged exposure to hypertonic solutions, and reduced post-thaw viability [9] [65].
- The Rationale for a Multi-Phasic Model: The Fast-Slow-Fast model is engineered to address these limitations:
- Initial Fast Phase: A rapid cooling rate from the starting temperature to just below the solution's freezing point minimizes the time cells spend in a supercooled state at high sub-zero temperatures, where chilling injury and cryoprotectant (CPA) toxicity can occur [7].
- Controlled Slow Phase: A subsequent slow, controlled cooling rate through the region of maximum ice formation facilitates sufficient cellular dehydration, preventing IIF. This is the critical phase for managing water efflux.
- Final Fast Phase: Once the cell suspension has achieved a high viscosity and the majority of freezable water has been removed, a rapid cooling rate to the final storage temperature is employed to vitrify the remaining intracellular solution, avoiding the destructive effects of ice recrystallization [65].
The following diagram illustrates the logical relationship between cooling rates and the biophysical phenomena they are designed to control.
Experimental Protocols & Data Analysis
Implementing the Fast-Slow-Fast model requires a controlled-rate freezer (CRF) and systematic experimentation to define the optimal parameters for a specific cell type.
Detailed Methodology for Protocol Optimization
A representative workflow for developing a Fast-Slow-Fast protocol is outlined below.
Key Considerations:
- Cryoprotectant Agent (CPA): The choice and concentration of CPA are integral to the model. For complex cells, a combination of permeating (e.g., Me₂SO) and non-permeating (e.g., hydroxyethyl starch, sugars) CPAs is often beneficial [43] [65]. For example, a study on rat mesenchymal stem cells found that a combination of 5% Me₂SO and 5% HES could be effective, reducing the required concentration of cytotoxic Me₂SO [43].
- Thawing: Thawing must be rapid to prevent ice recrystallization during warming. A warming rate of 45°C/min or higher is often recommended, typically achieved in a 37°C water bath or with a controlled-thawing device [7].
Quantitative Data and Comparative Analysis
The impact of different cooling rates on cell viability is well-documented. The table below summarizes quantitative findings from key studies.
Table 1: Comparative Analysis of Cooling Rate Impact on Cell Viability
| Cell Type | Cooling Rate Profile | CPA Formulation | Post-Thaw Viability / Outcome | Source |
|---|---|---|---|---|
| Rat MSCs | "Straight freeze" (passive) vs. Controlled -1°C/min | 5% Me₂SO / 5% HES | No significant difference in post-thaw viability between methods. | [43] |
| Mouse Subcutaneous Tissue | Slow Freezing | 10% Glycerinum | 26.0% of tissue blocks showed fibroblast growth. | [66] |
| Mouse Subcutaneous Tissue | Rapid Freezing | 10% Glycerinum | 6.7% of tissue blocks showed fibroblast growth. | [66] |
| Mouse Subcutaneous Tissue | Slow Freezing | 20% DMSO | No fibroblast growth observed. | [66] |
| Various (iPSC-derived, engineered) | Default CRF profile | Varies | Often suboptimal; requires profile optimization for sensitive types. | [7] |
Table 2: The Scientist's Toolkit: Essential Research Reagents and Materials
| Item | Function / Explanation | Example Application |
|---|---|---|
| Controlled-Rate Freezer (CRF) | Provides precise, programmable control over cooling rates for implementing complex multi-phasic profiles. | Essential for the Fast-Slow-Fast model; allows definition of each cooling segment [7]. |
| Cryoprotectants (Permeating) | Penetrate the cell membrane, providing intracellular protection by reducing ice crystal formation and stabilizing proteins. | Me₂SO (5-10%) is most common; Glycerol and 1,2-Propanediol are alternatives [43] [66] [65]. |
| Cryoprotectants (Non-Permeating) | Remain outside the cell, modifying extracellular ice formation and reducing osmotic shock. | Hydroxyethyl Starch (HES), sucrose, trehalose. Often used in combination with permeating CPAs [43] [65]. |
| Chemically Defined Cryomedium | Animal-component-free medium ensuring consistency, safety, and regulatory compliance for clinical applications. | Commercial formulations like CryoStor are designed to improve post-thaw recovery and reduce Me₂SO-related toxicity [67]. |
| Viability Assay Kits | Quantify post-thaw cell survival and function. Assessments should occur hours or days after thawing for accurate recovery data. | MTT assay, flow cytometry with Annexin V/PI, and Trypan Blue exclusion (e.g., performed 3 days post-thaw) [43] [67]. |
Discussion and Implementation
The data clearly indicates that a "one-size-fits-all" cooling rate is insufficient for the diverse landscape of complex cells. The success of the Fast-Slow-Fast model depends on customizing its parameters—the transition temperatures and the specific rates for each phase—to the specific cell type, its membrane permeability properties (Lp and Ps), and the CPA cocktail used [7] [65].
A significant challenge in the field is the qualification of CRFs and the definition of optimal profiles. Industry surveys indicate that while 60% of users start with a default CRF profile, many encounter challenges with sensitive cells like iPSC-derived lineages, necessitating customized optimization [7]. Furthermore, post-thaw analytics should extend beyond immediate viability to include long-term functionality, differentiation capacity, and potency assays to fully validate the protocol [9] [67].
The optimization of cooling rate profiles is a critical frontier in cryobiology, directly supporting the advancement of off-the-shelf cell therapies. The Fast-Slow-Fast model represents a sophisticated approach that acknowledges the dynamic nature of the freezing process. By systematically addressing the distinct biophysical risks present at different temperature stages, this model offers a pathway to significantly enhance the post-thaw recovery of delicate and therapeutically vital cells. Future work will involve deeper integration of thermodynamic modeling and real-time process analytics to further refine and standardize these protocols for robust clinical and commercial manufacturing.
Within the broader context of research on freezing rates and their profound impact on cell dehydration and ice formation, annealing emerges as a critical controlled interruption in the cooling process. This deliberate post-nucleation hold time at a specific sub-zero temperature is not a process of mere waiting; it is an active lever to manage the physical and osmotic dynamics of water within and outside the cell. The fundamental goal of cryopreservation is to avoid the lethal formation of intracellular ice (IIF), which is a primary cause of cell death [68] [13]. While rapid cooling risks IIF by trapping water inside the cell, slow cooling promotes protective dehydration but can lead to damaging "solution effects" due to prolonged exposure to high solute concentrations [69]. Annealing strategically balances these competing injuries. By holding the temperature after external ice has nucleated, annealing provides additional time for intracellular water to egress osmotically across the membrane before the cell cools to temperatures where IIF becomes imminent. This in-depth technical guide explores the mechanisms, protocols, and applications of annealing as a powerful tool for optimizing cell survival in cryopreservation and biopreservation.
Theoretical Foundation: Annealing in the Context of Freezing Injury
The Two Mechanisms of Freezing Injury
The response of cells to freezing is fundamentally governed by the cooling rate, which dictates the dominant pathway of injury:
- Slow Cooling Injury: At low cooling rates, cells are exposed to the external frozen environment for an extended period. Extracellular ice formation concentrates solutes in the unfrozen fraction, creating a steep osmotic gradient that draws water out of the cell. This leads to profound cellular dehydration and shrinkage. Damage occurs due to the prolonged exposure to high solute concentrations ("solution effects") and the potential for excessive shrinkage that can compromise the integrity of the cell membrane and internal structures [69] [13].
- Rapid Cooling Injury: At high cooling rates, the time allotted for water to leave the cell is insufficient. The cell's contents become increasingly supercooled, and eventually, the supercooling is terminated by the lethal formation of intracellular ice [68]. Intracellular ice crystals can physically disrupt organelles and the plasma membrane, leading to almost certain cell death.
The Role of Annealing in Modulating Water Transport
Annealing, or a post-nucleation hold time, is an interrupted cooling protocol that is strategically employed to mitigate rapid cooling injury [69]. The process is initiated once extracellular ice has formed (nucleation), ensuring the extracellular solution is frozen and the chemical potential of water outside the cell is lowered.
The subsequent hold at a temperature above the point where significant intracellular ice formation occurs provides a critical window for equilibration. During this hold time, the osmotic gradient, established by the extracellular ice, drives water out of the cell. This promotes protective dehydration, reduces the amount of supercooled water inside the cell, and thereby dramatically lowers the probability of IIF when cooling is resumed [69]. The annealing step effectively shifts the outcome of a moderately fast cooling protocol toward the more favorable dehydration pathway associated with slow cooling, without the associated time-dependent solution effects of very slow cooling.
Table 1: Key Cryobiological Concepts Relevant to Annealing
| Concept | Description | Relationship to Annealing |
|---|---|---|
| Intracellular Ice Formation (IIF) | The nucleation and growth of ice crystals inside the cell, which is almost always lethal [68]. | Annealing aims to prevent IIF by promoting cellular dehydration. |
| Supercooling | The state where water remains liquid below its equilibrium freezing point. | The driver for IIF; annealing reduces intracellular supercooling. |
| Osmotic Dehydration | The loss of water from a cell due to an osmotic gradient across its membrane. | This is the primary mechanism leveraged during the annealing hold time. |
| Cell Membrane Permeability | The rate at which water (and cryoprotectants) can cross the cell membrane. | Determines the kinetics of dehydration and thus the optimal duration of annealing. |
| Nucleation | The initial formation of an ice crystal, either extracellularly or intracellularly. | Annealing is a post-nucleation step, always initiated after extracellular ice is present. |
Quantitative Data on Annealing Effects
The effects of annealing are quantifiable and have been demonstrated across various biological systems, from individual cells to complex food and biological tissues.
Annealing in Cell Spheroids and Tissues
Cell-to-cell interactions significantly influence intracellular ice formation. Studies on hamster fibroblast models demonstrated that cells with extensive cell-cell contacts, such as those in multicellular spheroids, exhibit different IIF behavior compared to single cells in suspension. The temperature for intracellular freezing in 50% of the cells was significantly affected by these interactions, and there was evidence of intercellular nucleation through cell-cell junctions [70]. This highlights that in tissue-like structures, annealing must be designed to manage water transport across a coupled cellular network, not just individual membranes.
Furthermore, research on mouse oocytes and embryos revealed a dramatic shift in IIF temperature at the morula stage (compacted eight-cell embryo). While one-cell to eight-cell embryos underwent IIF at around -40°C, the nucleation temperature in early morulae was markedly higher, at approximately -23°C. This shift coincides with the formation of gap junctions and the expression of aquaporin 3 water channels, creating a functionally syncytial system that alters water transport and ice propagation dynamics [68]. Annealing protocols must therefore be tailored to the developmental and biological context, accounting for the presence of specialized membrane structures.
Annealing in Structural and Food Science Contexts
The principles of annealing extend beyond single-cell cryopreservation. In the freeze-drying of probiotic pellets, annealing is a critical step to modify the ice crystal structure formed during quenching in liquid nitrogen. Varying annealing durations directly influences the material thickness of the final freeze-dried product. Studies show that more extensive annealing results in thicker material, which exhibits a positive correlation with the storage stability of the encapsulated bacteria, particularly in oxygen-rich environments [71]. This demonstrates that annealing can be used to engineer protective microenvironments.
In food science, magnetic field-assisted osmotic dehydration (a process akin to annealing with an external field applied during the hold step) before freezing strawberries significantly improved outcomes. This pretreatment shortened freezing time, reduced water loss during thawing, and better preserved hardness and cell structure. The treatment resulted in a more uniform water distribution and better water retention, as measured by low-field NMR, underscoring how annealing can manage water to minimize structural damage [72].
Table 2: Summary of Quantitative Annealing Effects from Literature
| System | Annealing Protocol | Key Quantitative Outcome | Reference |
|---|---|---|---|
| Probiotic Pellets | Varying durations at -20°C or -9°C | Positive correlation between annealing-induced material thickness and storage stability (CFU counts). | [71] |
| Mouse Morulae | N/A (Study of intrinsic IIF temperature) | IIF nucleation temp. shifted from ~-40°C (one-cell) to -23.1 ± 1.5°C (early morula). | [68] |
| Strawberries | Osmotic dehydration assisted by magnetic field | Significantly reduced drip loss and malondialdehyde accumulation; improved hardness. | [72] |
| Hamster Fibroblasts | N/A (Study of cell interaction effects) | Cell-cell and cell-surface interactions significantly altered the temperature for IIF in 50% of cells. | [70] |
Experimental Protocols for Annealing
Designing an effective annealing protocol requires careful consideration of temperature, duration, and the cellular system's unique properties.
General Workflow for an Annealing Experiment
The following diagram outlines the key stages in a generalized interrupted cooling protocol that incorporates an annealing step.
Detailed Methodology
1. Sample Preparation:
- Cells: Prepare a suspension of the target cells at the desired concentration.
- Cryoprotective Agent (CPA): Prepare the freezing medium containing a permeating CPA (e.g., 1M Ethylene Glycol) and potentially non-permeating agents (e.g., sucrose). The CPA concentration and type are critical variables [68].
- Equilibration: Incubate the cell suspension with the CPA for a sufficient time (typically 10-15 minutes at room temperature or a defined chilling temperature) to allow permeation and osmotic equilibration [68].
2. Cooling to Nucleation Temperature:
- Cool the sample at a controlled rate (e.g., 1-10°C/min) using a programmable freezer or a pre-cooled alcohol bath to a temperature just below the freezing point of the medium.
- Induce Extracellular Nucleation: At approximately -5°C to -10°C, manually seed the sample by touching it with a pre-cooled instrument (e.g., forceps cooled in LN₂) or using an automated seeding function. This ensures controlled extracellular ice formation without significant intracellular supercooling.
3. Annealing Hold (The Critical Step):
- Temperature (T_anneal): Immediately after seeding, hold the sample at a defined annealing temperature. This is typically chosen to be within a range high enough to avoid significant IIF (e.g., -5°C to -20°C) but low enough to maintain a strong chemical potential gradient for water efflux. The optimal temperature is cell-type specific and depends on the membrane's water permeability and its temperature dependence.
- Duration (tanneal): Hold the sample at Tanneal for a predetermined time. This can range from several minutes (e.g., 5-10 min) to hours, depending on the cell type and the goals of the protocol. The duration must be sufficient to allow near-equilibration of water across the membrane [69]. For complex systems like probiotic pellets, annealing can be extended to hours or even days to coarsen the ice crystal structure for improved freeze-drying [71].
4. Resumption of Cooling:
- After the annealing hold, resume cooling at a controlled rate to the final temperature (e.g., -40°C to -80°C) before transfer to long-term storage in liquid nitrogen.
5. Thawing and Viability Assessment:
- Thaw samples rapidly (e.g., in a 37°C water bath) to avoid recrystallization of small intracellular ice crystals that may have formed.
- Perform viability assessments using standardized assays (e.g., membrane integrity stains like propidium iodide, functional assays, or clonogenic assays).
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Equipment for Annealing Experiments
| Item | Function / Relevance | Example / Specification |
|---|---|---|
| Permeating CPA | Penetrates cell membrane, depresses freezing point, reduces IIF risk. | Ethylene Glycol, Dimethyl Sulfoxide (DMSO), Glycerol [68] [73]. |
| Non-Permeating CPA | Creates osmotic gradient for dehydration, stabilizes membranes. | Sucrose, Trehalose, Hydroxyethyl Starch (HES) [71] [73]. |
| Programmable Freezer | Provides precise control over cooling rates, hold times, and temperatures. | Planer, Cryomed, or other brands with multi-segment programming. |
| Cryomicroscopy System | Allows direct visualization of ice formation, cell shrinkage, and IIF in real-time. | Linkam or other stages with temperature control and video recording [70] [68]. |
| Seeding Tool | To initiate controlled extracellular ice nucleation. | Pre-cooled metal forceps or an automated seeding probe. |
| Viability Assay Kits | To quantify post-thaw cell survival and function. | Flow cytometry kits (Annexin V/PI), metabolic activity assays (MTT), or clonogenic assays. |
| Low-Field NMR | To non-invasively analyze water status, distribution, and mobility in tissues or pellets. | Used to measure T2 relaxation times, indicating water retention [72] [13]. |
Mechanisms and Pathways: A Visual Synthesis
The protective effect of annealing is a consequence of coordinated physical and physiological events. The following diagram synthesizes the primary mechanism by which a post-nucleation hold time promotes cell survival.
Diagram: Mechanism of Annealing-Mediated Cell Protection. The process begins with (1) Extracellular Nucleation, which triggers (2) Establishment of an Osmotic Gradient. This gradient, maintained during the annealing hold, drives (3) Water Egress from the cell, leading to protective dehydration. The ultimate outcome is (4) a Reduced Risk of Lethal Intracellular Ice Formation and higher post-thaw survival.
Annealing, the strategic implementation of a post-nucleation hold time, is a powerful and versatile lever in the cryopreservation toolkit. Its role within freezing rate research is pivotal, as it effectively decouples the cooling rate from the time available for protective cellular dehydration. By providing a controlled window for osmotic water egress, annealing directly counteracts the primary mechanism of rapid cooling injury—intracellular ice formation. The success of an annealing protocol, however, is not universal; it is contingent on a deep understanding of the specific cellular system, including its membrane permeability, the presence of cell-cell junctions, and the composition of the extracellular medium. As cryobiology advances, the integration of annealing with novel polymeric cryoprotectants [73] and advanced physical modeling will continue to refine this technique, enabling the successful preservation of increasingly complex and sensitive biological systems for research and therapeutic applications.
Cryoconcentration, the uneven distribution of solutes within a frozen solution, presents a significant challenge in the biopharmaceutical industry, particularly for sensitive therapeutics like proteins and cell therapies. During freezing, the formation of ice crystals excludes solutes, leading to their concentration in the remaining liquid phase. This creates localized regions of high solute concentration, which can compromise critical product quality attributes [74]. For protein-based therapeutics, this process can induce aggregation, a major physical instability that may impact drug activity, solubility, and potentially trigger immunogenic responses [75]. In the context of cell therapies, such as CAR-T cells, the freezing process and the resulting osmotic imbalances can influence the efficacy, safety, and stability of the final product [4]. A deep understanding of the interplay between freezing rates, ice formation, and cellular dehydration is therefore fundamental to developing effective mitigation strategies for cryoconcentration within a broader research framework investigating the effects of freezing on cells.
Mechanisms of Cryoconcentration and Cell Dehydration
The process of freezing an aqueous solution, whether it contains proteins or living cells, initiates a series of complex physical events. As the temperature drops, water begins to form pure ice crystals externally. This ice formation excludes solutes, leading to a progressive increase in the concentration of dissolved substances in the remaining unfrozen liquid. This phenomenon, known as freeze-concentration or cryoconcentration, creates osmotic pressure gradients and can cause significant pH shifts in buffered solutions [75].
When the solution contains living cells, such as T-cells in therapy products, a critical osmotic imbalance occurs. The higher extracellular solute concentration draws intracellular water out of the cells, leading to cellular dehydration. If dehydration is insufficient, intracellular ice formation (IIF) can occur upon further cooling, which is typically lethal to cells as it disrupts cellular structures. Conversely, excessive water removal leads to a harmful increase in intracellular solute concentration, potentially damaging cellular machinery [4]. The cooling rate is a decisive factor in this balance; a slow cooling rate (approximately -1°C/min) is often used for mammalian cells to allow sufficient time for water to leave the cell and avoid IIF [4]. The following diagram illustrates the critical pathways and outcomes for a cell during a freeze-thaw cycle.
Quantitative Analysis of Freezing Parameters
The following tables consolidate key quantitative data from experimental studies on freezing processes, providing a reference for the critical parameters affecting cryoconcentration and cell viability.
Table 1: Impact of Cooling Rates on Cellular Outcomes
| Cooling Rate | Intracellular Dehydration | Intracellular Ice Formation (IIF) | Typical Application | Post-Thaw Viability Outcome |
|---|---|---|---|---|
| Slow (~ -1 °C/min) | Significant, controlled [4] | Minimal [4] | Mammalian cells, CAR-T cells [4] | High, if dehydration is not excessive [4] |
| Rapid (~ -10 °C/min or faster) | Insufficient [4] | High probability [4] | Not recommended for most cells | Low due to IIF [4] |
| Ultra-Rapid (e.g., Vitrification) | Minimal | Prevented | Specific sensitive cell types | Variable, requires high [4] CPA concentration |
Table 2: Effect of Controlled Ice Nucleation on Jurkat Cell Cryopreservation (Model for CAR-T Cells)
| Ice Nucleation Temperature | DMSO Concentration | Intracellular Dehydration | Intracellular Ice Formation | Post-Thaw Recovery |
|---|---|---|---|---|
| Spontaneous (Uncontrolled) | 2.5 - 5% (v/v) | Variable and insufficient | Higher incidence | Lower and more variable [4] |
| Controlled (Tnuc: -6 °C) | 2.5 - 5% (v/v) | Enhanced and more uniform | Significantly reduced | Improved [4] |
| Controlled (Tnuc: -10 °C) | 2.5 - 5% (v/v) | Less than Tnuc -6°C | Higher than Tnuc -6°C | Less improvement [4] |
| N/A (Control) | 10% (v/v) | N/A | N/A | High (Positive Control) [4] |
Table 3: Impact of Freeze-Thaw Rates on Monoclonal Antibody (mAb-1) Aggregation
| Freeze-Thaw Condition | Freeze Rate | Thaw Rate | Observed Aggregation | Recommendation |
|---|---|---|---|---|
| Slow Freeze - Fast Thaw | 0.03 °C/min [75] | 1 °C/min [75] | Minimized for mAb-1 [75] | Preferred condition for this mAb [75] |
| Fast Freeze - Slow Thaw | 1 °C/min [75] | 0.03 °C/min [75] | Significant increase [75] | Avoid for this mAb [75] |
Experimental Protocols for Characterization
A systematic approach to freeze-thaw characterization is imperative for identifying optimal conditions and mitigating cryoconcentration. The following protocols detail key methodologies.
Low-Temperature Thermal Analysis for Formulation Characterization
This protocol aims to determine the critical thermal properties of a protein formulation, such as its freezing point, glass transition temperature, and eutectic melt temperature, which are essential for designing a rational freeze-thaw cycle [75].
- Materials: Protein solution, electrical resistance meter, freeze-drying microscope with a temperature-controlled stage, Low-Temperature Differential Scanning Calorimeter (LT-DSC).
- Method:
- Resistance Analysis: Dispense the protein sample into a glass tube. Insert a ceramic resistance probe and a 32-gauge thermocouple. Cool and warm the sample at a controlled rate of 0.5 °C per minute. Record the temperature and electrical resistance every ten seconds. A deviation in resistance upon warming indicates the onset of the phase transition (eutectic melt) [75].
- Freeze-Drying Microscopy: Dispense a small volume of the protein solution into a glass cell on a freeze-drying stage equipped with thermocouples. Cool the sample at 0.5 °C/min to -60 °C and then warm at the same rate. Visually observe and record the sample's behavior (ice structure, collapse, melt-back) using a microscope with a camera [75].
- LT-DSC Analysis: Load 20-21 mg of the protein solution into a sample pan and crimp the lid. Purge the sample chamber with nitrogen. Cool the sample from room temperature to -65 °C and then warm at a controlled rate of 10 °C/minute. Monitor the heat flow to identify thermal events (e.g., crystallization, glass transition, melting) [75].
- Data Analysis: The combined data reveals the minimum temperature for complete solidification and the product's thawing characteristics, informing the target temperatures for storage and thawing [75].
Thin-Film Microscopy for Direct Observation of Ice Formation and Cell Dehydration
This protocol utilizes a specialized microscopy setup to directly visualize intracellular ice formation and cell volume changes in response to different freezing parameters, providing mechanistic insight [4].
- Materials: Cell suspension (e.g., Jurkat cells), cryoformulation with a fluorescent dye (e.g., acridine orange, propidium iodide), thin-film microscopy apparatus with polarized light and fluorescence capabilities, controlled rate freezer stage, pressure shift device for controlled nucleation (e.g., Control Lyo) [4].
- Method:
- Sample Preparation: Mix the cell suspension with the desired cryoformulation (e.g., varying DMSO concentrations in Plasma-Lyte A) and a fluorescent membrane integrity stain [4].
- Freezing Run: Place a small volume of the cell suspension as a thin film on the temperature-controlled microscope stage. Initiate a controlled cooling rate (e.g., -1 °C/min). For controlled nucleation experiments, initiate ice formation at a specific temperature (e.g., -6 °C) using a pressure shift method. For spontaneous nucleation, allow ice to form without intervention [4].
- Data Collection: During the freezing process, use polarized light to monitor ice crystal formation and fluorescence microscopy to track cell volume changes (dehydration) and the occurrence of intracellular ice formation (evidenced by sudden darkening of the cell). Continue imaging through the thawing phase to assess membrane integrity via fluorescence [4].
- Data Analysis: Quantify the incidence of IIF and the degree of cell dehydration under different nucleation temperatures and cooling rates. Correlate these observations with post-thaw viability measurements from bulk experiments.
The Scientist's Toolkit: Research Reagent Solutions
The following table lists key materials and reagents essential for conducting research into cryoconcentration and optimizing freeze-thaw protocols.
Table 4: Essential Research Reagents and Materials
| Item | Function/Application | Specific Examples / Notes |
|---|---|---|
| Controlled-Rate Freezer | Precisely controls cooling rate to minimize cryoconcentration and IIF; enables consistent, scalable processes [74] [4]. | Liquid nitrogen-free freezers are increasingly used [4]. |
| Single-Use Freeze-Thaw Bags | Container for drug substance; 2D geometry maximizes thermal transfer and reduces cryoconcentration compared to bottles [74]. | Aramus bags (high purity, non-leachable) [74]. |
| Cryoprotectants (CPAs) | Protect cells/proteins from freeze-thaw stresses; permeating CPAs reduce IIF, non-permeating CPAs mitigate osmotic shock. | DMSO (7-10% for cells) [4]; Sucrose, Trehalose (for proteins). |
| Stable Formulation Buffers | Maintain pH during freezing; prevents harmful pH shifts caused by cryoconcentration of buffer salts. | Histidine, Succinate; concentration and type require optimization [75]. |
| Surfactants | Mitigate protein aggregation at ice-water interfaces by reducing surface-induced denaturation [75]. | Polysorbate 20, Polysorbate 80. |
| Viability & Membrane Integrity Stains | Assess cell survival and membrane damage post-thaw in cell-based cryopreservation studies. | Acridine Orange, Propidium Iodide [4]. |
| Analytical Techniques for Aggregation | Quantify and monitor protein aggregation resulting from freeze-thaw stresses. | Size Exclusion HPLC (SE-HPLC), Analytical Ultracentrifugation (AUC) [75]. |
Strategic Workflow for Process Optimization
Implementing a structured workflow from small-scale modeling to scale-up is critical for successfully transferring a robust freeze-thaw process to manufacturing. The following diagram outlines this strategic approach.
In the realm of cell and gene therapy, cryopreservation serves as a pivotal bridge, enabling the storage and distribution of living cellular material across time and geography. While substantial research has focused on optimizing freezing rates to manage cell dehydration and ice formation, the thawing process has historically received less scientific scrutiny. This imbalance is particularly problematic given that thawing represents the final, critical barrier between a preserved cellular product and its therapeutic application. The process of rewarming is not merely a reversal of freezing; it introduces unique biophysical stresses that can independently compromise cell viability and function. Within the context of a broader thesis on the effect of freezing rates on cell dehydration and ice formation, this review establishes the thawing imperative—the undeniable scientific and clinical necessity to control warming parameters with the same precision as cooling protocols. The rate of warming and the concurrent management of osmotic shock are not merely procedural details but are fundamental determinants of final cell recovery, directly impacting the efficacy and consistency of cell-based therapies [7] [76].
This technical guide synthesizes current evidence to delineate how warming rate and osmotic stress interact to impact post-thaw outcomes. It provides drug development professionals and researchers with a structured framework for understanding thaw-induced injuries, alongside actionable data and protocols to optimize recovery of their most critical cellular assets.
The Biophysics of Thawing: From Glass Transition to Osmotic Shock
The Thermodynamic Race Against Ice Recrystallization
During cryopreservation, cellular solutions achieve a metastable state, either as a glassy solid (vitrification) or with a fraction of unfrozen water (slow freezing). The return to physiological temperatures is a race against thermodynamic instability. A primary threat during this phase is devitrification and ice recrystallization, which occur when the warming rate is insufficient to bypass the temperature zone (typically -50°C to -15°C) where ice crystals can melt, reorganize, and grow larger [76]. These larger crystals can exert mechanical forces that rupture plasma membranes and disrupt subcellular organelles [76] [13]. The required warming rate to prevent devitrification is often higher than the cooling rate required to achieve vitrification, due to the absorption of latent heat and the presence of myriad tiny ice embryos that can drive nucleation [76]. Rapid warming minimizes the time the sample spends in this dangerous temperature window, thereby preserving the amorphous or fine-crystalline state until the ice melts completely.
Navigating Osmotic Stress During Thawing
As ice melts, the extracellular environment transitions from a hypertonic, freeze-concentrated solution to an isotonic physiological medium. This shift subjects cells to profound osmotic stress. If thawing is uncontrolled, the rapid influx of water into dehydrated cells can cause excessive swelling and lysis [7] [4]. Furthermore, the dilution of cryoprotective agents (CPAs) like dimethyl sulfoxide (DMSO) must be managed. While DMSO is essential for preventing intracellular ice formation during freezing, it becomes cytotoxic at room temperature and can induce unwanted osmotic fluxes if not removed properly post-thaw [77] [78]. The optimal thawing protocol, therefore, must balance the need for rapid thermal warming to avoid ice recrystallization with the need for controlled osmotic compensation to prevent volume excursion damage.
Quantitative Evidence: Correlating Warming Rates with Cell Recovery
The relationship between warming rate and cell recovery is cell-type specific and influenced by the initial freezing protocol. The following table summarizes key quantitative findings from recent studies on different cell types.
Table 1: Impact of Warming Rate and Protocol on Post-Thaw Recovery of Different Cell Types
| Cell Type | Freezing Protocol | Warming Protocol | Key Outcome Metrics | Reference |
|---|---|---|---|---|
| T Cells (Jurkat model) | Slow cooling (-1°C/min) with 10% DMSO | Rapid warming in 37°C water bath | High viability post-thaw; Controlled nucleation at -6°C improved dehydration and reduced IIF. | [4] |
| Ovarian Tissue | Slow-freezing vs. Vitrification | "Universal" rapid warming protocol (~45°C/min) | Comparable or superior follicular count & reduced apoptosis vs. conventional thawing. | [79] |
| PBMCs | Slow freezing with 10% DMSO | Rapid warming in 37°C water bath | High viability & functionality maintained for up to 2 years with proper cryomedium. | [78] |
| Natural Killer (NK-92) | Standard freezing with DMSO | Rapid warming in 37°C water bath | Post-thaw viability 70-90%; recovery 30-80%; significant apoptosis (up to 84% loss in 24h). | [80] |
| C2C12 Myoblasts | Varying initial cooling rates (1-30°C/min) | Unified rapid thaw (50°C/min) | Slow cooling (1°C/min) before LN2 gave 65% viability vs. 54% for fast cooling (30°C/min). | [40] |
The data underscores that a rapid thermal warming rate is a near-universal requirement for high recovery. However, the table also highlights a critical interaction: the quality of the final thaw is contingent upon the initial freeze. For instance, the study on C2C12 myoblasts demonstrates that a slow initial cooling rate, which promotes better cell dehydration and accommodation within the freeze-concentrated solution (FCS), sets the stage for a more successful rapid thaw [40]. Conversely, a poor freezing process creates intracellular and extracellular ice architectures that are inherently more susceptible to damage during rewarming, a deficit that even an optimal warming rate cannot fully rectify.
Experimental Protocols for Assessing Thawing Outcomes
To systematically evaluate the impact of thawing parameters, researchers require robust and reproducible assays. Below is a detailed methodology for a foundational experiment comparing warming rates and osmotic shock on a given cell type.
Detailed Protocol: Warming Rate and Osmotic Stress Assay
Objective: To determine the optimal warming rate and post-thaw handling procedure for maximizing viability and functionality of a specific cell type (e.g., T cells, NK cells).
Materials:
- Cells: Target cell population (e.g., expanded T cells).
- Cryomedium: Standardized formulation (e.g., Plasma-Lyte A with 10% DMSO).
- Equipment: Controlled-rate freezer (CRF), water bath (37°C), dry bath, thermocouple data logger, centrifuge.
- Thawing Solutions: Pre-warmed complete culture medium, or a serial dilution of cryomedium.
- Viability/Functionality Assays: Flow cytometer with Annexin V/PI staining, cell counter (trypan blue), functional assay (e.g., cytokine secretion, cytotoxicity assay).
Methodology:
- Cell Freezing: Cryopreserve a homogeneous batch of cells using a standardized slow-freezing protocol (e.g., -1°C/min) in the CRF. Store all vials in the vapor phase of liquid nitrogen to ensure identical freezing history.
- Variable Thawing:
- Rapid Warm (Group A): Immediately transfer a cryovial from LN2 to a 37°C water bath with gentle agitation until only a small ice crystal remains.
- Slow Warm (Group B): Thaw a cryovial at ambient temperature (~22°C) or in a 4°C refrigerator, monitoring complete melt.
- Post-Thaw Processing:
- Direct Dilution (Subgroup 1): Immediately after thaw, transfer cell suspension directly into a 10x volume of pre-warmed complete medium.
- Controlled Dilution (Subgroup 2): After thaw, add pre-warmed medium drop-wise to the cell suspension over 1-2 minutes with gentle mixing, gradually diluting the DMSO before a final centrifugation and resuspension.
- Assessment:
- Viability & Recovery: Measure immediate post-thaw viability (Trypan Blue) and calculate percent recovery ((post-thaw viable cell count / pre-freeze viable cell count) * 100).
- Apoptosis: Use Annexin V/PI staining at 2-4 hours post-thaw to quantify early and late apoptosis.
- Functionality: Culture cells for 18-24 hours and then perform a functional assay (e.g., measure IFN-γ secretion upon stimulation).
This experimental workflow, which moves from a standardized freeze through systematic variations in thaw and post-thaw handling, can be visualized in the following diagram.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Thawing Studies
| Item | Function/Description | Example Application |
|---|---|---|
| Defined Cryopreservation Media | Serum-free, xeno-free media with known DMSO concentration; eliminates batch variability and ethical concerns of FBS. | Long-term storage of PBMCs for clinical trials [78]. |
| Cryoprotectant (DMSO) | Permeating agent that depresses freezing point and reduces intracellular ice formation; requires controlled removal post-thaw. | Standard cryopreservation of T cells and NK cells at 5-10% v/v [4] [80]. |
| Osmotic Buffers (e.g., Plasma-Lyte A) | Isotonic, pH-balanced solution used as a base for cryomedium or for post-thaw washing; minimizes non-osmotic chemical stress. | Formulating cryoprotectant solutions for Jurkat/T-cell models [4]. |
| Viability Stains (Trypan Blue, PI/Annexin V) | Dyes to discriminate live/dead cells (Trypan Blue, PI) and identify apoptotic cells (Annexin V). | Standard viability check post-thaw; detailed apoptosis analysis hours post-thaw [80] [40]. |
| Controlled-Rate Freezer (CRF) | Programmable freezer to apply precise, reproducible cooling profiles; essential for upstream process consistency. | Standardized freezing at -1°C/min for mammalian cells [7] [4]. |
Integrated Thawing Workflow and Damage Mechanisms
A comprehensive understanding requires viewing the thawing process as an integrated workflow, where thermal and osmotic stresses interact to dictate cellular fate. The following diagram maps the critical pathway from the frozen state to final recovery, highlighting key decision points and the mechanisms of damage that occur when protocols are suboptimal.
The mechanisms illustrated—ice recrystallization during slow warming and osmotic lysis during uncontrolled dilution—are not merely theoretical. They are observable and quantifiable. For example, the high post-thaw apoptosis in NK cells [80] and the reduced recovery of C2C12 myoblasts under suboptimal freezing conditions [40] are direct manifestations of these damage pathways. A successful protocol must therefore attack both fronts simultaneously.
The evidence is unequivocal: thawing is a deterministic process that demands deliberate scientific control. The "thawing imperative" mandates that warming rate and osmotic management be elevated from an afterthought to a critical process parameter in the development of cell-based therapeutics. The data shows that a rapid thermal warming rate is essential to circumvent the physical damage of ice recrystallization, while a controlled osmotic compensation strategy is equally vital to navigate the biochemical shock of cryoprotectant dilution.
Looking forward, the field must move beyond a one-size-fits-all approach. Future research should focus on cell-type specific optimization, recognizing that the biophysical properties of a mesenchymal stem cell differ from those of a CAR-T cell or an NK cell [80]. Furthermore, as the industry grapples with scale-up, the development of closed, automated thawing systems that reproducibly deliver both rapid thermal transfer and controlled dilution will be crucial for ensuring product quality and patient safety [7]. Finally, the exploration of novel CPA cocktails and natural cryoprotectants could potentially widen the therapeutic window for thawing, reducing the cytotoxicity burden and making the process more robust [77] [80]. By integrating a fundamental understanding of freezing-induced cell state with precision thawing, the next generation of cryopreservation protocols will significantly enhance the final recovery of viable, functional, and potent cellular products.
Benchmarking Success: Validation Techniques and Comparative Analysis of Freezing Regimens
The investigation of freezing rates on cell dehydration and ice formation is a critical area of research in biopreservation and pharmaceutical development. The cellular response to freezing is predominantly governed by the kinetics of water phase transitions, which directly impact cell viability and functionality. Within this research context, three analytical techniques form a complementary toolkit for systematic validation: Cryomicroscopy provides direct visual evidence of intracellular ice formation and cellular morphological changes, Differential Scanning Calorimetry (DSC) delivers quantitative thermal data on phase transitions and unfreezable water fractions, and Low-Field Nuclear Magnetic Resonance (LF-NMR) characterizes water state dynamics and mobility within biological systems. Together, these instruments enable researchers to correlate processing conditions with cellular outcomes, thereby elucidating the complex interplay between freezing parameters and biological responses. This technical guide examines the principles, methodologies, and integrated application of these tools within the specific context of freezing rate research, providing a comprehensive framework for experimental design and data interpretation.
Differential Scanning Calorimetry (DSC)
Principles and Applications
Differential Scanning Calorimetry operates on the principle of measuring heat flow into or out of a sample as a function of temperature or time. In freezing rate studies, DSC provides critical quantitative data on thermal transitions, including the enthalpy of fusion, glass transition temperatures, and the fraction of freezable versus unfreezable water within a biological matrix. This information is indispensable for constructing state diagrams, which map the physical states of a material as a function of both temperature and moisture content [81]. For instance, DSC has been instrumental in determining that canola seeds exhibit a critical moisture content threshold of approximately 16%, below which all remaining water exists as unfreezable water [81]. Furthermore, the technique can identify multiple glass transition events, as observed in canola seeds where the second and third glass transition temperatures decreased to 63.46°C and 73.38°C, respectively, at high moisture contents [81]. These measurements provide fundamental insights into how water plasticizes biological systems at different freezing temperatures.
Experimental Protocol for Freezing Studies
Sample Preparation: For biological cell suspensions, prepare samples in relevant cryoprotectant solutions (e.g., 2.5-5% DMSO in Plasma-Lyte A for T-cells) [27]. For plant or food materials, whole seeds or tissue sections of consistent mass (4-6 mg) may be used directly [81]. Seal samples in hermetic aluminum pans to prevent moisture loss during analysis.
DSC Procedure: Utilize a cool-hold-heat method with controlled nitrogen purge gas (flow rate 50 mL/min). Begin with an isothermal hold at 25°C for 5 minutes, then cool to -80°C at a controlled rate (e.g., 1°C/min for high sensitivity to thermal events), hold for 5 minutes, and finally heat to 25°C at the same rate [81]. Multiple cooling and heating rates (1, 2, 5, 10, and 20°C/min) may be tested to determine kinetic effects.
Data Analysis: Determine the freezing point from the exothermic peak temperature during cooling. Identify glass transition temperatures (Tg) from the inflection point in the baseline shift during heating. Calculate freezable water content from the enthalpy of melting (ΔH) using the relationship: % Freezable Water = (ΔHsample/ΔHpure water) × 100, where ΔHpure water = 334 J/g [81].
Table 1: DSC Thermal Parameters in Freezing Research
| Parameter | Description | Research Significance | Exemplary Data |
|---|---|---|---|
| Freezing Point Depression | Temperature where exothermic freezing initiates | Indulates solute effect and critical moisture content | -26°C at 17.8% MC in canola [81] |
| Glass Transition (Tg) | Temperature range where amorphous matrix transitions from glassy to rubbery state | Predicts storage stability and molecular mobility | Multiple Tg events observed in canola seeds [81] |
| Freezable Water Fraction | Percentage of water that undergoes crystallization | Determines cellular dehydration extent and ice formation risk | Critical MC of 16% in canola seeds [81] |
| Melting Enthalpy | Energy absorbed during ice melting | Quantifies ice content and crystal size distribution | Varies with MC and thermal history [81] |
Low-Field Nuclear Magnetic Resonance (LF-NMR)
Principles and Applications
Low-Field NMR relaxometry probes the molecular dynamics of water in biological systems by measuring the relaxation times of hydrogen protons following radiofrequency excitation. The spin-spin relaxation time (T2) is particularly informative, as it distinguishes between water populations based on their mobility: bound water (short T2), immobilized water (intermediate T2), and free water (long T2) [82]. In freezing research, LF-NMR tracks how water redistribution and phase changes correlate with freezing damage. Studies on frozen gel models have demonstrated that freezing temperatures significantly impact T2 distributions, with lower temperatures shifting populations toward shorter relaxation times, indicating restricted water mobility [82]. The bi-exponential character of spin-spin relaxation processes in tissues further provides insights into the microenvironments of water in different cellular compartments [83]. When combined with deep learning algorithms, LF-NMR data can predict post-thaw quality attributes such as drip loss and texture with high accuracy, establishing it as a powerful non-destructive validation tool [82].
Experimental Protocol for Freezing Studies
Sample Preparation: For cell suspensions or tissues, load 2-3 g into standard NMR tubes. For model systems, gelatin-water mixtures (e.g., 1:9 and 2:8 ratios) provide standardized matrices [82]. Subject samples to defined freezing protocols at temperatures ranging from -5°C to -80°C.
LF-NMR Procedure: Utilize a Fast Field Cycling (FFC) NMR relaxometer to perform measurements across a frequency range from 10 kHz to 10 MHz [83]. For T2 relaxation measurements, employ the Carr-Purcell-Meiboom-Gill (CPMG) pulse sequence with sufficient echoes (typically 1000-5000) to capture the full decay. Maintain a constant temperature during measurement using a precision temperature controller.
Data Analysis: Invert CPMG decay data to obtain T2 distributions using algorithms such as CONTIN or T-invfit software [82]. Integrate peak areas to quantify water populations. For frequency-dependent spin-lattice relaxation (R1), fit data to power-law functions (R1(ω) = Cω^(-α)) to elucidate water dynamics mechanisms [83]. Employ machine learning models (e.g., Back-Propagation Artificial Neural Networks) to correlate LF-NMR parameters with quality attributes.
Table 2: LF-NMR Parameters in Freezing Research
| Parameter | Description | Research Significance | Exemplary Data |
|---|---|---|---|
| T2 Relaxation Time | Time constant for spin-spin relaxation | Distinguishes bound, immobilized, and free water states | Three distinct peaks in frozen gel models [82] |
| T2 Peak Area Ratio | Relative proportion of different water populations | Quantifies water redistribution during freezing | Correlates with drip loss in thawed samples [82] |
| Power-law Exponent (α) | Parameter describing frequency dependence of R1 | Reveals mechanisms of water dynamics (e.g., reptation) | α ≈ 0.3 in lung tissues, indicating polymer-like dynamics [83] |
| Bi-exponential T2 Components | Fast and slow relaxing fractions in tissues | Differentiates intra- and extracellular water environments | Marked differences between tumor and healthy lung tissue [83] |
Cryomicroscopy
Principles and Applications
Cryomicroscopy encompasses several techniques for imaging frozen specimens at cryogenic temperatures. Traditional cryogenic electron microscopy (cryo-EM) has been revolutionized by recent advances that address the challenge of imaging thick biological samples. A key development is tilt-corrected bright-field scanning transmission electron microscopy (tcBF-STEM), which places the imaging optics before the sample, thereby eliminating chromatic blurring from inelastic scattering that plagues conventional TEM in thick specimens [84] [85]. This approach achieves a 3-5× improvement in dose efficiency compared to energy-filtered TEM, enabling high-contrast imaging of intact bacterial cells and large organelles up to 500-800 nm thick [84]. For studying dynamic freezing processes, cryomicroscopic setups allow direct observation of intracellular dehydration and ice formation in real-time [27]. These studies have revealed that controlled ice nucleation at higher temperatures (e.g., -6°C) promotes greater intracellular dehydration and reduces detrimental intracellular ice formation, correlating with improved post-thaw T cell viability [27].
Experimental Protocol for Freezing Studies
Sample Preparation: For cellular studies, Jurkat T-cells in cryoprotectant solutions (e.g., DMSO in Plasma-Lyte A) are loaded into specialized cryo-stages [27]. For structural studies, vitrify samples by plunge-freezing in liquid ethane and mount on EM grids [85].
Imaging Procedure: For dynamic freezing studies, use a cryomicroscopic stage with controlled temperature ramp. Initiate freezing with controlled ice nucleation at defined temperatures (e.g., -6°C vs. -10°C) and monitor cellular responses in real-time [27]. For structural tcBF-STEM: cool the sample to cryogenic temperatures; acquire 4D-STEM datasets with a defocused probe using an electron microscope pixel array detector (EMPAD); raster the probe across the sample while recording 2D diffraction patterns at each position [84] [85].
Data Analysis: For dynamic studies, quantify intracellular ice formation versus dehydration by tracking changes in cell volume and optical properties [27]. For tcBF-STEM: correct image shifts between images formed from individual detector pixels; combine shift-corrected images to generate a final high signal-to-noise ratio image [84]. Perform single-particle analysis for structural determination of macromolecular complexes.
Integrated Approach and Research Reagent Solutions
Correlative Methodology
The most powerful insights emerge from integrating these analytical techniques rather than using them in isolation. For instance, DSC can identify thermal transition temperatures that guide cryomicroscopy experimental parameters, while LF-NMR can quantify water states that correlate with microscopic ice crystal observations. A compelling example of integration is found in the combined use of cryo-EM and solid-state NMR to investigate protein fibril structures, where NMR provides atomic-level secondary structure information and cryo-EM delivers detailed morphological context [86]. Similarly, in freezing rate studies, DSC determines the critical temperature thresholds for ice formation, cryomicroscopy visually confirms the location and extent of this ice formation, and LF-NMR characterizes the resulting water mobility changes in different cellular compartments. This tripartite approach enables researchers to establish mechanistic links between processing conditions, water state transitions, and cellular outcomes.
Research Reagent Solutions
Table 3: Essential Research Reagents and Materials
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Cryoprotective agent | Prevents intracellular ice formation in T-cell cryopreservation [27] |
| Plasma-Lyte A | Isotonic carrier solution | Base medium for cryopreservation formulations [27] |
| Gelatin | Model food matrix | Creates standardized gel models for freezing studies [82] |
| Petroleum Ether | Lipid solvent | Extracts oil from seeds for controlled DSC studies [81] |
| EMPAD Detector | Electron detection | Enables 4D-STEM data acquisition in tcBF-STEM [84] [85] |
| Jurkat T-Cells | Model cell line | Study intracellular dehydration and ice formation [27] |
Experimental Workflows and Data Interpretation
Integrated Workflow for Freezing Rate Studies
The following diagram illustrates the logical relationship between experimental techniques and the specific parameters they measure in a comprehensive freezing rate study:
Data Correlation and Interpretation
Successful interpretation of freezing rate studies requires cross-correlation of data from all three analytical platforms. For example, when DSC detects a higher freezable water fraction at a specific cooling rate, cryomicroscopy might visually confirm larger extracellular ice crystals, while LF-NMR could show a corresponding increase in the more mobile water population (longer T2 component). Conversely, optimized freezing protocols that maintain cell viability typically show: minimal intracellular ice formation (cryomicroscopy), a significant fraction of unfreezable water (DSC), and a water state profile dominated by restricted mobility components (LF-NMR). Researchers should develop correlation matrices that quantitatively link parameters across techniques, enabling predictive models for cell survival based on measurable physical parameters. This integrated analytical approach provides the comprehensive validation framework necessary to optimize cryopreservation protocols in pharmaceutical development and cellular therapeutics.
The interdisciplinary application of DSC, LF-NMR, and cryomicroscopy provides an unparalleled analytical framework for investigating the complex interplay between freezing rates, water state transitions, and cellular outcomes. DSC delivers essential quantitative data on thermal transitions and freezable water fractions; LF-NMR characterizes water dynamics and mobility non-destructively; and advanced cryomicroscopy techniques like tcBF-STEM enable high-resolution visualization of ice formation in thick biological specimens. The integration of these methodologies allows researchers to move beyond correlative observations toward mechanistic understandings of freezing-induced cellular damage. As these analytical technologies continue to evolve—particularly with improvements in detector sensitivity, computational analysis, and correlative approaches—they will undoubtedly yield deeper insights into the fundamental processes of ice formation and cell dehydration, ultimately enabling more effective preservation strategies for biological systems in pharmaceutical and clinical applications.
The cryopreservation of cells is a cornerstone of modern biotechnology, regenerative medicine, and drug development. The core challenge lies in mitigating the inherent damage caused by the freezing process, which directly impacts post-thaw cell quality and functionality. The effect of freezing rates on cell dehydration and ice formation represents a critical area of research, as these physical phenomena are primary determinants of cryoinjury. Controlled-rate freezing aims to manage water transport across the cell membrane, minimizing lethal intracellular ice formation (IIF) by promoting protective cell dehydration [87]. However, insufficient or excessive dehydration can lead to different damage pathways. This technical guide provides researchers with a contemporary framework for quantitatively measuring the triad of essential post-thaw outcomes—viability, membrane integrity, and functionality—enabling the critical evaluation of cryopreservation protocols within this fundamental biophysical context.
Core Mechanisms: Freezing Rates, Dehydration, and Ice Formation
The freezing rate is a master variable that dictates the dominant pathway of water transport and ice formation, creating a delicate balance between two major mechanisms of cryoinjury.
- Slow Freezing Rates promote osmotic dehydration. As extracellular ice forms, solutes concentrate in the unfrozen fraction, creating an osmotic gradient that draws water out of the cell. This is protective against IIF, but excessive dehydration can lead to "solution effects," including damaging changes in pH, solute concentration, and macromolecular structure, and can cause irreversible membrane fusion and shrinkage [87].
- Rapid Freezing Rates risk intracellular ice formation. When the cooling rate is too fast, water does not have sufficient time to exit the cell before the intracellular contents supercool and nucleate ice. The formation of intracellular ice crystals is almost universally lethal, causing mechanical disruption of organelles and the plasma membrane [88] [40].
The morphological features of the freeze-concentrated solution (FCS)—the network of channels where cells and solutes are sequestered between ice crystals—are also governed by the freezing rate. Slow cooling (~1°C/min) produces relatively large FCS channels, whereas rapid cooling (10-30°C/min) results in finer ice crystals and narrower FCS channels, which can impede cell accommodation and reduce the protective effect of cryoprotectants [40]. This is quantified in Table 1, which summarizes the relationship between cooling rate and key biophysical parameters.
Table 1: Impact of Initial Cooling Rate on Cryopreservation Outcomes
| Initial Cooling Rate | Extracellular Ice Crystals | FCS Channel Width | Cell Dehydration | Intracellular Ice Formation | Typical Cell Recovery |
|---|---|---|---|---|---|
| Slow (~1°C/min) | Large, dendritic | Larger | Significant | Minimal | Higher (e.g., ~65%) [40] |
| Medium | Mixed morphology | Moderate | Moderate | Moderate | Variable/Variable [40] |
| Rapid (10-30°C/min) | Small, numerous | Narrower | Insufficient | Extensive | Lower (e.g., 54-59%) [40] |
The formation and subsequent recrystallization of ice during thawing can cause significant mechanical damage to cellular structures. As shown in a 2025 model using frozen meat, ice crystals damage muscle cells, promote protein/lipid oxidation, and reduce water-holding capacity [13]. In cell systems, this directly translates to compromised membrane integrity and functionality.
Measuring Post-Thaw Outcomes: A Multi-Parameter Approach
A comprehensive assessment of cryopreservation success requires a multi-faceted approach that moves beyond simple viability to include membrane integrity and critical cellular functions.
Viability and Membrane Integrity
Viability and membrane integrity are most commonly assessed using dye-based assays that distinguish between live and dead/damaged cells based on plasma membrane permeability.
- Acridine Orange (AO) / Propidium Iodide (PI) or 7-AAD Staining: AO is a membrane-permeant green nucleic acid stain that labels all cells. PI or 7-AAD are membrane-impermeant stains that only enter cells with compromised plasma membranes, binding to DNA and labeling them red (PI) or fluorescent in the far-red spectrum (7-AAD). Viability is calculated as the percentage of AO-positive cells that exclude PI/7-AAD. A 2025 clinical study on hematopoietic stem cells found AO staining to be highly sensitive for detecting delayed post-thaw degradation [89].
- Trypan Blue Exclusion: This is a long-standing, simple method where the blue dye is excluded by live cells with intact membranes but accumulates in dead cells. It is often used with a hemocytometer for cell counting and viability assessment post-thaw [88] [40].
Table 2: Key Assays for Post-Thaw Assessment
| Assessment Category | Specific Assay | Measured Parameter | Technical Note |
|---|---|---|---|
| Viability / Membrane Integrity | AO/PI or 7-AAD Flow Cytometry | Plasma membrane integrity | Considered a gold standard; allows for high-throughput analysis [89]. |
| Trypan Blue Exclusion | Plasma membrane integrity | Simple, rapid; can be combined with automated cell counters [88]. | |
| Cellular Function & Metabolism | Colony-Forming Unit (CFU) Assay | Clonogenic capacity / proliferative potential | Critical for stem cells; indicates long-term functional health [90]. |
| Metabolic Activity (e.g., CCK-8) | Overall metabolic function | Measures enzymatic activity; reflects population health [40]. | |
| Lineage-Specific Function | Differentiation Assays | Post-differentiation phenotype & markers | e.g., THP-1 to macrophages; flow cytometry for CD14/CD11b [88]. |
| CD34+ Cell Dose & Engraftment | Functional potency in vivo | Key clinical correlate for hematopoietic stem cells [89]. |
Cellular Functionality
Functionality assays are essential, as a viable cell with an intact membrane may not possess its full native biological activity.
- Clonogenic Capacity: The Colony-Forming Unit (CFU) assay is a robust method for assessing the proliferative potential of stem and progenitor cells. Post-thaw cells are plated in semi-solid media and monitored for their ability to form distinct colonies over 1-2 weeks. This is a stringent test of functional fitness, as used in evaluations of cord blood mononuclear cells [90].
- Metabolic Activity: Assays like the Cell Counting Kit-8 (CCK-8), which measures the reduction of a WST-8 tetrazolium salt to a colored formazan product by cellular dehydrogenases, provide a snapshot of the population's metabolic health post-thaw [40].
- Lineage-Specific Differentiation: For cells like the THP-1 monocyte line, the ultimate functional test is their ability to differentiate into target cells (e.g., macrophages) after thawing. This is assessed by morphological changes (shift from round to ameboid, adherent cells) and upregulation of differentiation markers (e.g., CD14, CD11b) via flow cytometry. A 2025 study showed that optimized cryopreservation with macromolecular cryoprotectants enabled post-thaw differentiation capacity comparable to non-frozen controls [88].
The diagram below illustrates the logical workflow for designing experiments and interpreting the relationships between freezing conditions, cellular damage, and the resulting measurable outcomes.
Diagram: The logical relationship between freezing parameters, biophysical events, and post-thaw measurable outcomes. The balance managed by cooling rate and cryoprotectants (CPAs) directly influences the extent of dehydration and ice formation, which in turn dictates the severity of cryoinjury reflected in key assays.
Advanced Protocols for Key Experiments
Protocol: Cryopreservation of Sensitive Cell Lines (e.g., THP-1 Monocytes) with Advanced Cryoprotectants
This protocol, adapted from a 2025 study, demonstrates how macromolecular cryoprotectants can enhance post-thaw recovery and functionality for difficult-to-preserve immune cells [88].
- Cell Preparation: Culture THP-1 cells in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, and 1% antibiotic-antimycotic. Maintain cells in exponential growth phase.
- Cryomedium Formulation: Prepare cryopreservation medium consisting of base culture medium (RPMI-1640 with 2 mM L-glutamine) supplemented with:
- 20% (v/v) Fetal Bovine Serum (FBS)
- 5% (v/v) DMSO
- 40 mg/mL synthetic polyampholyte (e.g., polymer with mixed cationic/anionic side chains). Sterile-filter the final solution using a 0.22 µm syringe filter.
- Freezing Procedure:
- Centrifuge cells at 100 RCF for 5 minutes. Resuspend the pellet in the cryomedium to a density of 1 × 10^6 viable cells/mL.
- Aliquot 1 mL of cell suspension into cryovials.
- Place cryovials in an isopropanol-based freezing container (e.g., CoolCell) and transfer immediately to a -80°C freezer for at least 24 hours. For long-term storage, transfer vials to liquid nitrogen after 24 hours.
- Thawing and Assessment:
- Rapidly thaw cryovials in a 37°C water bath for approximately 2 minutes.
- Dilute the thawed cell suspension 1:10 with pre-warmed thawing medium (RPMI-1640 with 20% FBS, 2 mM L-glutamine, 1% antibiotic-antimycotic).
- Centrifuge at 100 RCF for 5 minutes, decant the supernatant, and gently resuspend the cell pellet in fresh culture medium.
- Perform cell counts and viability assessment using trypan blue exclusion or AO/PI flow cytometry.
- Functional Differentiation Assay:
- Plate the thawed and recovered THP-1 cells in tissue culture plates.
- Induce macrophage differentiation by adding 100 ng/mL Phorbol 12-myristate 13-acetate (PMA) to the culture medium for 48 hours.
- Assess differentiation success by:
- Microscopy: Observe morphological change from round, suspension cells to large, ameboid, adherent cells.
- Flow Cytometry: Detect upregulation of macrophage surface markers (e.g., CD14 and CD11b).
Protocol: Analysis of Freeze-Concentrated Solution (FCS) Morphology
This morphological study links the physical freezing environment to cell recovery [40].
- Sample Preparation: Prepare a 5-10% (v/v) DMSO solution in water or buffer. Add a fluorescent dye like sodium fluorescein (100 µM) to visualize the liquid phase.
- Freezing and Imaging:
- Place a 10 µL aliquot of the solution between two microscope slides on a precision cooling stage.
- Program the stage to cool at specific rates (e.g., 1°C/min, 10°C/min, 30°C/min).
- Use fluorescence microscopy with a 4x objective to capture images of the forming FCS channels during the freezing process.
- Image Analysis:
- Use image analysis software (e.g., ImageJ) to measure the width of the FCS channels.
- Analyze a minimum of 40 non-overlapping channels per condition.
- Generate histograms of channel width and fit with Gaussian functions to compare the average size and distribution under different cooling rates.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Cryopreservation Research
| Item | Function / Application | Example / Note |
|---|---|---|
| Permeating CPA | Penetrates cell, reduces intracellular ice formation. | Dimethyl Sulfoxide (DMSO); use at 5-10% [40] [91]. |
| Macromolecular CPA | Extracellular; mitigates osmotic shock, inhibits ice recrystallization. | Synthetic polyampholytes; hydroxyethyl starch; dextran [88] [91]. |
| Ice Nucleator | Controls nucleation temperature, reduces supercooling & variability. | Pollen-derived extracts; specific proteins [88]. |
| Viability Stain | Distinguishes live/dead cells based on membrane integrity. | Acridine Orange/Propidium Iodide (AO/PI); 7-AAD; Trypan Blue [88] [89]. |
| Cell Recovery Assay | Measures metabolic activity as a proxy for viability/health. | Cell Counting Kit-8 (CCK-8) [40]. |
| Functional Assay Kits | Assesses clonogenic potential of stem/progenitor cells. | Colony-Forming Unit (CFU) methylcellulose media [90]. |
| Differentiation Inducers | Tests functional capacity of progenitor cell lines post-thaw. | Phorbol 12-myristate 13-acetate (PMA) for THP-1 cells [88]. |
| Controlled-Rate Freezer | Standardizes cooling protocol, critical for reproducibility. | Alternative: Isopropanol-based freezing containers for -80°C [88]. |
The rigorous measurement of post-thaw viability, membrane integrity, and cellular functionality is non-negotiable for advancing cryopreservation science. As research continues to elucidate the intricate relationships between freezing rates, cell dehydration, and ice formation, the assays and protocols outlined in this guide provide a pathway to data-driven optimization. By employing a multi-parameter assessment strategy that includes clonogenic and differentiation assays, researchers can move beyond simple viability to ensure that cryopreserved cells are not only alive but fully functional, thereby enhancing the reliability and success of downstream applications in therapy and drug development.
Cryopreservation is an indispensable technology in the field of cell therapy, influencing the safety, reliability, and effectiveness of cell-based products for patient recipients [4]. For T-cell therapies, including Chimeric Antigen Receptor (CAR)-T cells, the freezing process presents a critical manufacturing hurdle that can significantly impact product quality and therapeutic efficacy [4]. During freezing, intracellular events such as dehydration and ice formation directly influence post-thaw viability and functionality [27].
The ice nucleation temperature (Tn), defined as the temperature at which ice crystals first form in the solution, represents a critical process parameter in cryopreservation protocols [92]. This case study investigates how controlled manipulation of ice nucleation temperature affects intracellular dehydration, intracellular ice formation (IIF), and ultimately, the viability of Jurkat cells as a model T-cell system [4]. The findings are framed within the broader context of thesis research on how freezing rates influence cell dehydration and ice formation, providing technical guidance for researchers and drug development professionals seeking to optimize cryopreservation protocols for cell therapy products.
Theoretical Background: Ice Nucleation and Cellular Response
Fundamental Cryoinjuries in Cell Freezing
During cryopreservation, cells face two primary mechanical stresses as temperatures decrease:
- Extracellular Ice Formation: Ice forms initially in the extracellular solution, increasing solute concentration in the remaining liquid and creating an osmotic gradient that drives water out of cells [4].
- Intracellular Dehydration vs. Intracellular Ice Formation (IIF): The rate of cooling determines the balance between these competing outcomes. Slow cooling permits sufficient time for cellular dehydration, minimizing IIF but potentially increasing solute damage. Rapid cooling traps water inside cells, leading to lethal IIF [4].
The Critical Role of Ice Nucleation Temperature
Ice nucleation temperature directly influences the ice crystal structure and subsequent cellular responses. Spontaneous (uncontrolled) ice nucleation occurs stochastically at variable supercooled temperatures, leading to product heterogeneity [4]. Controlled ice nucleation allows initiation of freezing at a defined, higher temperature closer to the solution's equilibrium freezing point [4] [92].
The degree of supercooling (ΔT), calculated as the difference between the equilibrium freezing point and the actual ice nucleation temperature, governs ice crystal formation dynamics [92]:
Higher supercooling (lower Tn) results in more numerous, smaller ice crystals, increasing resistance to vapor flow during drying and creating complex pore structures [92]. For cellular systems, this translates to varied pathways of cellular dehydration and IIF risk.
Table 1: Fundamental Relationships in Ice Nucleation Physics
| Parameter | Definition | Impact on Freezing Process |
|---|---|---|
| Equilibrium Freezing Point | Temperature at which liquid and solid phases coexist in equilibrium | Solution property dependent on cryoprotectant type and concentration |
| Ice Nucleation Temperature (Tₙ) | Actual temperature at which ice crystals first form | Determines degree of supercooling; controllable via nucleation methods |
| Degree of Supercooling (ΔT) | Difference between equilibrium freezing point and Tₙ | Higher ΔT produces more ice nuclei, smaller crystals, increased product resistance |
The following diagram illustrates the conceptual relationship between ice nucleation temperature and the two competing pathways of cellular dehydration and intracellular ice formation:
Experimental Design and Methodology
Cell Model and Cryoformulations
This case study employs Jurkat cells (Clone E6-1, ATCC TIB-152) as a model T-cell line for CAR-T cell cryopreservation [4]. Cells were cultured in RPMI 1640 Medium supplemented with fetal bovine serum under standard conditions.
Cryoformulations consisted of Plasma-Lyte A with Dimethyl Sulfoxide (DMSO) at three concentrations:
- Low DMSO: 2.5% v/v DMSO in Plasma-Lyte A
- Medium DMSO: 5% v/v DMSO in Plasma-Lyte A
- High DMSO: 10% v/v DMSO in Plasma-Lyte A (positive control)
Ice Nucleation Experimental Conditions
The experimental design compared three ice nucleation conditions:
- High Tn: Controlled ice nucleation at -6°C (closer to equilibrium freezing point)
- Low Tn: Controlled ice nucleation at -10°C (further from equilibrium freezing point)
- Spontaneous Tn: Uncontrolled ice nucleation (stochastic supercooling)
Controlled ice nucleation was achieved using a pressurization/depressurization method (Control Lyo) as an alternative to liquid nitrogen-based ice fog techniques [4].
Analytical Techniques and Monitoring
A comprehensive analytical approach was employed to characterize cellular responses:
- Thin-Film Cryomicroscopy: Combined polarized light with fluorescence capabilities to dynamically monitor ice formation, cell volume changes, and membrane integrity during freeze-thaw processes [4].
- Bulk Freeze-Thaw Validation: Cells frozen in bulk using liquid nitrogen-free controlled rate freezer with post-thaw viability assessment via membrane integrity stains (acridine orange and propidium iodide) [4].
- Differential Scanning Calorimetry (DSC): Determined equilibrium freezing temperatures of cryoformulations and established DMSO/Plasma-Lyte A liquidus curve [4] [12].
Table 2: Key Experimental Parameters and Methodologies
| Experimental Component | Specifications | Application in Study |
|---|---|---|
| Cell Model | Jurkat cells (Clone E6-1), 1 × 10⁷ cells/mL | Representative T-cell model for CAR-T cryopreservation |
| Cryoprotectant | DMSO at 2.5%, 5%, 10% v/v in Plasma-Lyte A | Standard cryoprotectant at clinically relevant concentrations |
| Nucleation Methods | Pressurization/depressurization (Control Lyo) | Controlled nucleation without liquid nitrogen |
| Cooling Rate | Slow cooling (-1°C/min) followed by faster cooling (-10°C/min) below glass transition | Standard protocol for mammalian cell cryopreservation |
| Analytical Methods | Thin-film cryomicroscopy, fluorescence staining, DSC | Multi-parameter assessment of cellular responses |
Results and Discussion
Impact of Nucleation Temperature on Cellular Dehydration and Ice Formation
Cryomicroscopy studies revealed significant differences in cellular responses based on nucleation temperature:
- High Tn (-6°C): Promoted more intracellular dehydration with less incidence of intracellular ice formation during freezing compared to either lower nucleation temperature or uncontrolled nucleation [4] [27].
- Low Tn (-10°C) and Spontaneous Tn: Resulted in less dehydration and higher probability of intracellular ice formation, evident by darkening of cellular appearance indicating IIF [4].
These observations demonstrate that initiating ice formation at a temperature closer to the equilibrium freezing point allows sufficient time for water egress from cells before the system reaches temperatures where IIF becomes probable (typically below -20°C). The reduced supercooling associated with higher Tn creates a less steep chemical potential gradient, moderating the rate of water efflux and minimizing simultaneous ice formation inside and outside cells.
Membrane Integrity and Post-Thaw Viability
Bulk cryopreservation experiments consistently demonstrated the advantage of higher ice nucleation temperature:
- Cells frozen with Tn of -6°C showed significantly better membrane integrity post-thaw compared to other nucleation conditions across all DMSO concentrations tested [4].
- The correlation between observed cellular events (dehydration and IIF) during cryomicroscopy and final cell viability confirmed the mechanistic relationship between nucleation temperature and cryopreservation outcomes [27].
Notably, the benefits of controlled nucleation at -6°C were observed even at reduced DMSO concentrations (2.5% and 5%), suggesting that optimized nucleation protocols may enable formulation improvements for clinical applications where DMSO toxicity raises concerns [4].
Interaction Between Cooling and Warming Rates
Complementary research on human peripheral blood T cells has revealed important interactions between cooling and warming rates [12]:
- With slow cooling rates (-1°C/min or slower), warming rate had minimal impact on viable cell number across a wide range (1.6°C/min to 113°C/min) [12].
- Only with rapid cooling (-10°C/min) did slow warming rates (1.6°C/min and 6.2°C/min) reduce viable cell numbers, with cryomicroscopy revealing ice recrystallization during slow thawing that mechanically disrupted frozen cells [12].
These findings establish an operational envelope for T-cell cryopreservation, emphasizing that the benefits of controlled nucleation at higher temperatures are maximized when paired with appropriate cooling rates.
Table 3: Quantitative Outcomes of Ice Nucleation Temperature on T-Cell Cryopreservation
| Experimental Condition | Intracellular Dehydration | Intracellular Ice Formation | Membrane Integrity | Overall Viability |
|---|---|---|---|---|
| Tn -6°C (High) | Significantly enhanced | Substantially reduced | Best preservation | Highest recovery |
| Tn -10°C (Low) | Moderate | Increased compared to Tn -6°C | Reduced preservation | Lower recovery |
| Spontaneous Tn | Variable, generally limited | Highest incidence | Poor preservation | Lowest and most variable |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials and Reagents for Controlled Ice Nucleation Research
| Item | Function/Application | Specific Examples |
|---|---|---|
| Cell Model | Representative T-cell system for cryopreservation studies | Jurkat cells (Clone E6-1, ATCC TIB-152) [4] |
| Cryoprotectant | Prevents intracellular ice formation, reduces osmotic stress | DMSO at 2.5-10% v/v in Plasma-Lyte A [4] |
| Controlled Nucleation Device | Initiates ice formation at defined temperatures | Pressurization/depressurization systems (Control Lyo); Ice fog generators [4] [92] |
| Cryomicroscopy System | Real-time monitoring of ice formation and cellular responses | Thin-film microscopy with polarization and fluorescence [4] |
| Viability Assays | Quantifies post-thaw cell survival and function | Membrane integrity stains (acridine orange, propidium iodide) [4] |
| Thermal Analysis | Characterizes thermal properties of cryoformulations | Differential Scanning Calorimetry (DSC) [4] |
Industry Context and Implementation Challenges
Recent industry surveys by the ISCT Cold Chain Management & Logistics Working Group highlight critical implementation considerations:
- Adoption Rates: 87% of survey participants use controlled-rate freezing for cell-based products, with only 13% relying on passive freezing (primarily in early clinical stages) [7].
- Qualification Challenges: 30% of respondents rely on vendors for system qualification, with limited consensus on qualification methodologies for controlled-rate freezers [7].
- Scale-Up Hurdles: 22% of industry professionals identify "Ability to process at large scale" as the biggest hurdle for cryopreservation in cell and gene therapy [7].
The following workflow summarizes the complete experimental process from cell preparation to data analysis in controlled ice nucleation studies:
This case study demonstrates that controlled ice nucleation at a defined higher temperature (-6°C) closer to the equilibrium freezing point of cryoformulations significantly improves T-cell cryopreservation outcomes by enhancing cellular dehydration while minimizing intracellular ice formation. The correlation between observed cellular events during freezing and post-thaw viability confirms the critical importance of nucleation control as a process parameter in cell therapy manufacturing.
For researchers and drug development professionals, these findings suggest that implementing controlled ice nucleation protocols could enhance product consistency and potentially enable reduced DMSO concentrations in final formulations. Future work should focus on scaling these approaches for commercial manufacturing and extending optimization to more complex cell therapy products, including primary CAR-T cells and other immune effector cells.
As the cell therapy industry advances with the market for cell freezing media projected to grow at a CAGR of 8.6% from 2025 to 2035 [93] [94], refining fundamental cryopreservation processes through mechanistic understanding of parameters like ice nucleation temperature will be essential for delivering consistent, high-quality therapeutic products to patients.
The cryopreservation of biological materials—from single cells to complex tissues—is fundamental to modern biomedical research, drug development, and clinical applications. The freezing rate stands as a critical determinant of post-thaw viability and functionality, directly influencing cellular responses to ice formation and osmotic stress. This technical guide provides a comprehensive analysis of slow and fast freezing methodologies, examining their differential effects across biological systems within the context of cell dehydration and ice formation dynamics. Understanding these mechanisms is paramount for developing optimized cryopreservation protocols that maintain structural integrity and biological function after thawing.
The core challenge in cryopreservation lies in managing the phase change of water. When biological samples freeze, extracellular ice formation initiates a cascade of physicochemical events. The equilibrium freezing point of aqueous solutions is approximately 0°C, but solutions commonly supercool to between -15°C and -60°C before ice nucleation occurs [44]. This extracellular ice formation concentrates solutes in the remaining liquid, creating an osmotic gradient that drives cellular dehydration—a process critical to preventing lethal intracellular ice formation (IIF) [4] [35].
Table 1: Fundamental Cryoinjuries Associated with Freezing Processes
| Cryoinjury Type | Primary Mechanism | Most Affected by |
|---|---|---|
| Intracellular Ice Formation (IIF) | Physical damage to membranes and organelles by internal ice crystals | Fast cooling rates |
| Excessive Dehydration | Osmotic water efflux causing irreversible cell shrinkage and solute damage | Slow cooling rates |
| Oxidative Stress | Reactive oxygen species (ROS) generation impairing cellular functions | Both processes, during freeze-thaw cycles |
| Cryoprotectant Toxicity | Chemical toxicity of protective agents (e.g., DMSO) | Concentration and exposure time |
Theoretical Foundations: Freezing Rate, Dehydration, and Ice Formation
The Two-Factor Hypothesis and Cellular Response
The theoretical framework for understanding freezing damage is largely built upon Mazur's "two-factor hypothesis," which posits that cellular injury during cryopreservation results from two primary mechanisms: intracellular ice formation at high cooling rates and excessive dehydration or "solute effects" at low cooling rates [1]. The interplay between cooling rate and cellular dehydration governs the choice between slow and fast freezing protocols for different biological systems.
During slow freezing, extracellular ice formation creates a chemical potential difference that drives water out of cells, leading to progressive dehydration. This dehydration concentrates intracellular solutes and cryoprotective agents (CPAs), thereby depressing the intracellular freezing point and reducing the probability of IIF. The rate of water efflux must be balanced against the cooling rate; if cooling is too rapid, water cannot exit cells quickly enough, resulting in supercooling and eventual intracellular freezing [55]. Conversely, excessively slow cooling prolongs exposure to hypertonic conditions, potentially causing toxicity and shrinkage-induced damage.
Advanced Modeling of Freeze-Thaw Processes
Recent computational models have enhanced our understanding of cellular behavior during cryopreservation. Advanced numerical approaches now simultaneously consider transmembrane transport of water and cryoprotectants, intracellular crystallization during cooling, and recrystallization during rewarming. These models reveal that higher cooling rates increase intracellular ice volume and elevate intracellular nucleation temperature [55].
For mouse oocytes, modeling determined that cooling rates of 0.5°C·min⁻¹ to 1°C·min⁻¹ cause minimal harm, while rewarming rate significantly influences intracellular ice volume—faster warming results in smaller increases in ice volume during the recrystallization phase [55]. Furthermore, models suggest that cryopreservation temperature should remain below -160°C with retrieval operations completed within 90 seconds to minimize damaging recrystallization events [55].
Comparative Experimental Data Across Biological Systems
Mammalian Spermatozoa
A controlled study comparing rapid freezing (RF) and slow programmable freezing (SPF) of human spermatozoa demonstrated clear differential outcomes. Post-thaw sperm motility was significantly higher in RF (53.9%) compared to SPF (37.0%), though both remained lower than non-frozen controls (75.5%) [95]. Similarly, sperm vitality assessed by hypo-osmotic swelling test was 60.1% for RF versus 44.1% for SPF (controls: 77.9%), and by eosin-Y staining was 64.8% for RF versus 50.4% for SPF (controls: 81.8%) [95].
Notably, no significant differences emerged in post-thaw normal sperm morphology (14.9% RF, 14.4% SPF, 16% controls) or DNA integrity assessed by comet assay (93.6% RF, 94.5% SPF, 94.2% controls) [95]. The hemi-zona index, measuring sperm binding capacity, showed no difference between cryopreserved groups, indicating that while motility parameters favored RF, fundamental functional integrity was preserved similarly by both methods.
Ovarian Tissue and Complex Biosructures
Ovarian tissue cryopreservation presents unique challenges due to its complex multicellular architecture. A study of bovine ovarian cortex fragments compared slow freezing and vitrification (an ultra-rapid cooling process) against fresh control tissue [96]. Morphological analysis of 1,344 follicles without cultivation and 552 with cultivation revealed no significant differences in non-atretic follicle counts between control (572), vitrification (289), and slow freezing (373) groups [96].
Following 7 days of culture, follicular development progression was observed across all groups, supporting that morphologically identified non-atretic follicles in non-cultivated groups were indeed viable [96]. The study concluded that with no protocol superiority established, vitrification represents an advanced alternative method particularly valuable for patients requiring fertility preservation due to its lower cost, faster processing, and better adaptability to laboratory routines [96].
T-Cells and Therapeutic Applications
Cryopreservation of cell therapy products like T-cells requires exceptional viability and functional recovery. Research investigating controlled ice nucleation in Jurkat cells (a T-cell model) revealed that ice nucleation temperature significantly impacts intracellular events [4]. Cryomicroscopic studies demonstrated that a higher ice nucleation temperature (-6°C), closer to the equilibrium freezing point of cryoformulations, promoted intracellular dehydration and reduced intracellular ice formation compared to lower nucleation temperatures (-10°C) or uncontrolled nucleation [4].
This enhanced dehydration profile correlated with improved cell membrane integrity and post-thaw viability in bulk cryopreservation, highlighting the importance of controlling early freezing events beyond merely setting cooling rates [4]. The findings have direct implications for manufacturing chimeric antigen receptor (CAR)-T cell therapies, where cryopreservation occurs not only for final products but also for intermediate manufacturing steps.
Molecular and Cellular Models
Cooling rate dependence was further elucidated using Chinese hamster ovary (CHO) cells overexpressing aquaporin-4 (AQP4) water channels [1]. At slow freezing rates (<35°C/min) with Me₂SO as CPA, viability remained high and stable, while trehalose (a non-permeating CPA) resulted in decreased viability [1]. However, at rapid cooling rates (>80°C/min), AQP4-expressing cells showed significantly higher viability than non-expressing cells, demonstrating that enhanced water permeability enables better survival under fast-freezing conditions [1].
Critically, no successful cryopreservation occurred without CPAs, even in AQP4-overexpressing cells with heightened water permeability [1]. This underscores that membrane water permeability alone is insufficient for cryoprotection and that cryoprotective agents remain essential for managing both dehydration and ice formation processes.
Table 2: Optimal Cooling Rates and Outcomes Across Biological Systems
| Biological System | Slow Freezing Rate | Rapid Freezing Rate | Key Outcome Measures | Preferred Method |
|---|---|---|---|---|
| Human Spermatozoa | Slow programmable freezing | Rapid freezing | Post-thaw motility: 37.0% (SPF) vs 53.9% (RF) [95] | Rapid freezing |
| Bovine Ovarian Tissue | Conventional slow freezing | Vitrification | Non-atretic follicles: 373 (SF) vs 289 (Vit) [96] | Equivalent outcomes |
| Jurkat T-Cells | ~1°C/min with controlled nucleation | Ultra-rapid with high CPA | Membrane integrity and dehydration management [4] | Slow freezing with controlled nucleation |
| CHO Cells | <35°C/min | >80°C/min | Viability maintenance with Me₂SO at slow rates [1] | Cell-type dependent |
| C2C12 Myoblasts | 1°C/min | 10-30°C/min | Cell recovery: 65% (slow) vs 54-59% (fast) [20] | Slow freezing |
The Role of Cryoprotective Agents and Supporting Technologies
Cryoprotectant Mechanisms and Limitations
Cryoprotective agents (CPAs) are essential components of both slow and fast freezing protocols, acting through multiple mechanisms to mitigate freezing damage. Permeating CPAs like dimethyl sulfoxide (DMSO) and glycerol enter cells and form hydrogen bonds with intracellular water, reducing ice crystal formation and growth by replacing water molecules in hydration shells [4] [35]. Non-permeating CPAs like trehalose and sucrose remain extracellular, creating osmotic gradients that promote protective dehydration before freezing and moderating water influx during thawing [1].
However, CPAs present significant challenges, particularly concentration-dependent toxicity. DMSO induces epigenetic alterations in hepatic and cardiac cells, inhibits osteoclast formation, and causes dramatic changes in cellular processes [4] [35]. Glycerol can cause hemolysis and alter red blood cell morphology [35]. These toxicities necessitate careful optimization of CPA concentration and exposure time, followed by post-thaw removal procedures that themselves can cause osmotic stress.
Ice Management Strategies
Advanced ice management strategies have emerged to improve cryopreservation outcomes. Controlled ice nucleation, where the extracellular ice formation temperature is precisely defined, demonstrates significant benefits for T-cell preservation [4]. By initiating ice formation at temperatures close to the solution's equilibrium freezing point (approximately -6°C), intracellular dehydration is enhanced while intracellular ice formation is reduced compared to spontaneous nucleation at lower temperatures [4].
Ice-binding proteins (IBPs), including antifreeze proteins and ice-nucleating proteins, offer another strategic approach. These proteins interact with ice crystal surfaces, modifying their formation and growth [44]. When incorporated into cryopreservation protocols, IBPs can inhibit recrystallization during thawing, potentially reducing cryoinjury and improving post-thaw recovery.
Experimental Protocols and Methodologies
Standardized Slow Freezing Protocol for Mammalian Cells
The following protocol is adapted from methodologies successfully employed for T-cells, ovarian tissue, and other mammalian cell systems [4] [96]:
- Pre-freeze Processing: Prepare cells through density gradient centrifugation or enzymatic dissociation as appropriate. Resuspend in growth medium containing 10% serum substitute.
- CPA Addition: Gradually add pre-chilled cryopreservation medium containing 10% DMSO to achieve final concentration, using stepwise addition to minimize osmotic shock.
- Packaging: Aliquot 1 mL of cell suspension (approximately 1-5×10⁶ cells/mL) into cryovials.
- Programmed Freezing:
- Place vials in controlled-rate freezer pre-cooled to 4°C.
- Cool at -1°C/min to -7°C.
- Hold for 5-10 minutes to ensure thermal equilibrium.
- Initiate ice nucleation (seeding) at -7°C when applicable.
- Continue cooling at -1°C/min to -40°C.
- Increase cooling rate to -10°C/min to -90°C.
- Transfer to liquid nitrogen vapor phase for long-term storage.
Rapid Freezing Protocol for Spermatozoa
This protocol is adapted from studies demonstrating superior post-thaw motility with rapid freezing techniques [95]:
- Semen Processing: Layer washed, density gradient-processed spermatozoa over cushion media.
- CPA Equilibration: Mix processed sperm with cryoprotectant solution containing glycerol and egg yolk extender to final concentration of 5-7%.
- Packaging: Load 0.5 mL aliquots into pre-chilled straws.
- Freezing Process:
- Place straws on rack 3-4 cm above liquid nitrogen surface for 10 minutes.
- Alternatively, use pre-programmed freezer with rapid cooling rate (>50°C/min).
- Plunge into liquid nitrogen for storage after initial freezing phase.
Vitrification Protocol for Ovarian Tissue
Adapted from successful bovine ovarian tissue cryopreservation studies [96]:
- Tissue Preparation: Cut ovarian cortex into fragments of 1 × 1 × 0.5 mm.
- CPA Equilibration:
- 3 minutes in 1.2 M glycerol solution
- 3 minutes in 1.2 M glycerol + 3.6 M ethylene glycol solution
- 1 minute in 3 M glycerol + 4.5 M ethylene glycol solution
- Cooling and Storage:
- Place individual fragments on aluminum foil strips
- Immediately immerse in liquid nitrogen
- Transfer to cryovials for storage at -196°C
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Cryopreservation Research
| Reagent/Material | Function | Example Applications |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating cryoprotectant; reduces intracellular ice formation | Standard cryopreservation of mammalian cells [35] |
| Glycerol | Permeating cryoprotectant; protects membrane integrity | Spermatozoa cryopreservation [95] |
| Trehalose | Non-permeating cryoprotectant; osmotic regulation | Alternative CPA in CHO cells [1] |
| Ethylene Glycol | Permeating cryoprotectant; rapid penetration | Vitrification solutions [96] |
| Plasma-Lyte A | Isotonic base solution; cryoformulation component | T-cell cryopreservation [4] |
| Hydroxyethyl Starch | Non-permeating CPA; extracellular protection | Red blood cell cryopreservation [20] |
| Antifreeze Proteins | Ice recrystallization inhibition; crystal morphology control | Enhanced cryopreservation outcomes [20] |
| Sucrose | Osmotic buffer; reduces osmotic shock during CPA removal | Standard component of thawing solutions [96] |
| Programmable Freezer | Controlled cooling rate apparatus; protocol standardization | Slow freezing applications [95] [96] |
| Cryomicroscopy System | Direct visualization of ice formation and cell response | Protocol development and optimization [4] |
The comparative analysis of slow versus fast freezing methodologies reveals that optimal cryopreservation outcomes depend on a complex interplay between biological system characteristics, cooling rate, cryoprotectant selection, and ice management strategies. No universal superior method exists; rather, the choice between slow and fast freezing approaches must be tailored to specific cellular properties and functional requirements.
Key determinants include cell surface-to-volume ratio, membrane water permeability, tolerance to osmotic stress, and sensitivity to cryoprotectant toxicity. Slow freezing generally favors systems where controlled dehydration prevents intracellular ice formation, while rapid freezing benefits systems requiring minimal exposure to concentrated solutes or those with naturally high water permeability.
Future advancements will likely emerge from integrated approaches combining optimized thermal protocols with advanced cryoprotectant formulations and ice-modulating agents. Such multidisciplinary strategies will enhance our ability to preserve increasingly complex biological systems, from therapeutic cell products to tissues and organs, supporting continued progress in regenerative medicine, pharmaceutical development, and fundamental biological research.
Designing Small-Scale Freeze-Thaw Studies to Predict Large-Scale Performance
The transition from small-scale experimentation to large-scale bioproduction represents a critical juncture in the development of cryopreservation protocols for cell therapies and biopharmaceuticals. This technical guide establishes a framework for designing small-scale freeze-thaw studies that accurately predict large-scale performance, specifically contextualized within pioneering research on the effects of freezing rates on cell dehydration and ice formation. The fundamental challenge lies in the cryodamage mechanisms—intracellular ice formation during rapid cooling versus excessive cellular dehydration and solute toxicity during slow cooling—first articulated in Mazur's "two-factor hypothesis" [1]. At scale, these phenomena are magnified by increased volume and complexity, making predictive modeling essential for success.
The core premise of this guide is that effective scale-translation requires more than simple volumetric increase; it demands a mechanistic understanding of ice crystallization kinetics and mass transport phenomena across different systems. By grounding small-scale experimental design in robust thermodynamic principles and cell-scale biophysics, researchers can create predictive models that ensure consistent cell viability, functionality, and recovery upon scaling to manufacturing volumes.
Scientific Foundation: Freezing Rate Effects on Cellular Responses
The Two-Factor Hypothesis and Its Implications
The conceptual foundation for freeze-thaw protocol design rests on Mazur's two-factor hypothesis, which describes the competing injury mechanisms during freezing:
Slow Freezing Damage: At suboptimal slow cooling rates, extracellular ice formation initiates a sequence of deleterious events. As extracellular water freezes, the unfrozen fraction becomes increasingly concentrated with solutes, creating an osmotic gradient that draws water out of cells. This causes profound cellular dehydration and exposure to hypertonic solutions, leading to solute-effect damage or "solution effects" that denature proteins and disrupt membrane integrity [1].
Rapid Freezing Damage: At supraoptimal rapid cooling rates, intracellular water has insufficient time to efflux across the membrane in response to extracellular ice formation. This results in intracellular supercooling followed by lethal intracellular ice formation (IIF), which mechanically disrupts organelles and membrane structures [1] [97].
The relationship between cooling rate and cell survival follows a classic inverted-U profile, with the optimal rate representing the balance between dehydration and intracellular ice formation. This balance point varies significantly between cell types based on their biophysical properties, particularly membrane water permeability and surface-to-volume ratio [55] [1].
Advanced Modeling of Freeze-Thaw Processes
Contemporary models extend beyond the original two-factor hypothesis to incorporate more complex phenomena:
Coupled Transport Models: Advanced numerical models now simulate the coupled transport of water and cryoprotectant agents (CPAs) across cell membranes during freezing, accounting for the non-ideal behavior of concentrated solutions and their impact on osmotic responses [55].
Intracellular Crystallization Dynamics: State-of-the-art models predict not only initial ice formation but also recrystallization during rewarming, a significant contributor to cryoinjury that was neglected in earlier models. These models can simulate how intracellular ice volume evolves throughout the entire freeze-thaw cycle [55].
Thermodynamic Parameters: Key thermodynamic properties including glass transition temperature (Tg'), crystallization temperature (Tc), and melting temperature (Tm) provide critical insights into system behavior during phase transitions and can be experimentally determined using differential scanning calorimetry [98].
Table 1: Critical Thermodynamic Parameters in Freeze-Thaw Process Design
| Parameter | Definition | Experimental Determination | Significance in Protocol Design |
|---|---|---|---|
| Glass Transition Temperature (Tg') | Temperature at which the maximally concentrated solution vitrifies | Differential Scanning Calorimetry (DSC) | Determines minimum storage temperature; defines the endpoint for slow cooling phase |
| Crystallization Temperature (Tc) | Temperature at which ice crystallization initiates during cooling | DSC | Informs seeding strategies and cooling rate optimization |
| Melting Temperature (Tm) | Temperature at which ice completely melts during warming | DSC | Guides thawing protocol design and defines complete melting point |
| Nucleation Temperature (Tn) | Actual temperature at which ice nucleation occurs | Visual observation or thermal monitoring | Affects ice crystal size and distribution; subject to stochastic variation |
Small-Scale Study Design Fundamentals
Core Experimental Parameters and Monitoring Approaches
Well-designed small-scale studies systematically evaluate critical parameters that govern freezing outcomes:
Cooling Rate Optimization: The cooling rate represents the primary experimental variable, with typical studies exploring a range from 0.3°C/min to >50°C/min. Different cell types demonstrate markedly different optimal cooling rates based on their membrane permeability characteristics [1]. For example, aquaporin-overexpressing cells maintain high viability at rapid cooling rates (-120°C/min) due to enhanced water efflux capacity, while conventional cells require much slower rates [1].
Nucleation Control: The stochastic nature of ice nucleation presents a significant scaling challenge. Small-scale studies should implement controlled nucleation techniques (e.g., manual seeding with chilled forceps, ultrasound, or ice-nucleating agents) at consistent supercooling thresholds (typically -5°C to -7°C) to improve inter-experimental consistency and scaling predictability [99].
CPA Selection and Toxicity Kinetics: The choice between permeating (DMSO, glycerol) and non-permeating (trehalose, sucrose) CPAs significantly impacts osmotic responses during freezing. Studies must evaluate both concentration-dependent cytotoxicity and time-dependent toxicity, as DMSO exposure duration directly correlates with cell damage [100] [99]. Recent approaches combine CPA types; for instance, using 1.5M DMSO with 0.1M sucrose has demonstrated efficacy for ovarian tissue cryopreservation [98].
Container Scale-Down Strategy
A critical principle in predictive small-scale study design is the implementation of a container scale-down strategy that maintains key aspects of the large-scale system:
Table 2: Container Scale-Down Strategy for Freeze-Thaw Studies
| Scale | Typical Volume | Recommended Container | Heat Transfer Characteristics | Scale-Relevant Considerations |
|---|---|---|---|---|
| Micro-scale | 0.1-0.5 mL | Cryovials (1.0-1.8 mL) | Rapid temperature equilibration | Useful for initial cooling rate screening |
| Bench-scale | 1-10 mL | Cryobags (5-25 mL) or larger vials | Moderate thermal mass | Closer approximation to manufacturing conditions |
| Pilot-scale | 10-100 mL | Laboratory-scale bags or bottles | Significant thermal lag | Essential for validating scaling parameters |
| Production-scale | 100-1000+ mL | Manufacturing-scale bags or bottles | Substantial thermal gradients | Reference system for scaling correlations |
Quantitative Parameters for Predictive Modeling
Cell-Scale Response Metrics
Small-scale studies must capture quantitative parameters that describe cell-level responses to freezing stresses:
Cell Volume Excursions: Monitoring volumetric changes during freezing provides insights into membrane permeability properties and osmotic tolerance limits. Measurements typically utilize computer-assisted sperm analysis (CASA) systems or image analysis of adherent cells [99].
Intracellular Ice Formation (IIF) Incidence: Quantifying IIF through cryomicroscopy or indirect measures provides critical data on the incidence of lethal freezing events across different cooling rates [55].
Post-Thaw Viability and Functionality: Comprehensive assessment extends beyond membrane integrity (typically measured via dye exclusion) to include metabolic activity, apoptosis incidence, and cell-specific functional assays that more accurately predict performance in downstream applications [99].
System Thermodynamic Metrics
The thermodynamic behavior of the freezing system provides essential scaling parameters:
Solution Characteristics: The combination of Leibovitz L-15 medium with 4 mg/mL human serum albumin (HSA), 1.5M DMSO, and 0.1M sucrose presents characteristic thermal transitions at Tg' = -120.49°C, Tc = -20°C, and Tm = -4.11°C when cooled at 2.5°C/min [98].
Ice Crystal Morphology Analysis: Crystal size distribution and fractal dimension analysis provide quantitative measures of ice structure that correlate with cell damage. Rapid freezing typically produces small, uniform crystals while slow freezing creates larger, more dendritic structures [13].
Temperature Profiles in Actual Containers: Monitoring thermal profiles at multiple locations within samples reveals the magnitude of thermal gradients that develop during scaling. These gradients significantly impact freezing kinetics and must be characterized to enable predictive modeling [55].
Experimental Protocols for Predictive Small-Scale Studies
Protocol 1: Cooling Rate Optimization Study
This protocol systematically characterizes the cooling rate dependence of specific cell types to identify optimal parameters:
Materials and Reagents:
- Cell suspension (≥90% viability pre-freeze)
- Cryoprotectant solution (e.g., culture medium with 10% DMSO)
- Controlled-rate freezer or passive freezing device
- Cryovials (1.0-1.8 mL)
- Water bath (37°C) for thawing
- Viability assessment reagents (e.g., trypan blue, flow cytometry reagents)
Procedure:
- Prepare cell suspension at standardized concentration (e.g., 1×10^7 cells/mL)
- Mix with equal volume of 2× CPA solution to achieve final concentration
- Aliquot 1.0 mL into cryovials and equilibrate for 15 minutes on ice
- Program controlled-rate freezer with following segment sequence:
- 4°C to -7°C at 1°C/min
- Hold at -7°C for 2 minutes, initiate seeding
- -7°C to -40°C at variable rates (0.3, 1, 5, 10, 20, 50°C/min)
- -40°C to -140°C at 10°C/min
- Transfer to liquid nitrogen storage
- After 24-72 hours, thaw rapidly in 37°C water bath with gentle agitation
- Assess viability immediately post-thaw and after 2-hour recovery
- Plot viability versus cooling rate to identify optimum
Data Interpretation: The optimal cooling rate typically falls at the apex of the viability versus cooling rate curve. Additional analysis should evaluate correlations between cooling rate and intracellular ice formation incidence, as well as functionality metrics where applicable [1].
Protocol 2: Thermodynamic Parameter Determination
This protocol characterizes the thermal transition properties of cryopreservation solutions to inform protocol design:
Materials and Reagents:
- Cryopreservation solution (identical composition to freezing medium)
- Differential scanning calorimeter (DSC)
- Hermetic DSC pans
- Reference empty pan
- Liquid nitrogen cooling system
Procedure:
- Precisely load 5-10 mg of solution into hermetic DSC pan
- Seal pan according to manufacturer specifications
- Program DSC method:
- Equilibrate at 25°C
- Cool to -150°C at 2.5°C/min
- Hold for 5 minutes
- Heat to 25°C at 5°C/min
- Analyze thermograms to identify:
- Glass transition temperature (Tg') - midpoint of transition
- Crystallization temperature (Tc) - peak of exotherm
- Melting temperature (Tm) - peak of endotherm
- Repeat with three separate samples for statistical reliability
Data Interpretation: Tg' defines the minimum practical storage temperature; Tc informs appropriate seeding temperatures; Tm guides thawing endpoint determination [98].
Protocol 3: Scale-Down Container Validation
This protocol validates that small-scale containers appropriately mimic large-system behavior:
Materials and Reagents:
- Cell suspension
- Cryopreservation medium
- Multiple container types/volumes (vials, bags)
- Multiple temperature loggers (where feasible)
- Controlled-rate freezer
Procedure:
- Prepare identical cell suspensions in cryopreservation medium
- Aliquot into different container types at varying volumes (0.5 mL, 2 mL, 5 mL, 10 mL)
- Place temperature probes in center of largest containers if feasible
- Subject all containers to identical freezing protocol
- Assess post-thaw viability and functionality for each container type/volume
- Compare outcomes across container configurations
Data Interpretation: Small-scale containers that produce comparable results to larger systems while demonstrating similar thermal profiles provide predictive capability for scaling [55].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Freeze-Thaw Studies
| Category | Specific Examples | Function and Application | Key Considerations |
|---|---|---|---|
| Permeating CPAs | Dimethyl sulfoxide (DMSO), Glycerol, Ethylene glycol | Penetrate cell membranes, reduce intracellular ice formation, depress freezing point | Concentration-dependent toxicity; DMSO most common but affects epigenetics [100] [97] |
| Non-Permeating CPAs | Trehalose, Sucrose, Hydroxyethyl starch | Create hypertonic extracellular environment, promote dehydration, stabilize membranes | Work osmotically; require membrane transporters for cellular uptake [1] |
| Ice Regulating Molecules | Antifreeze proteins (AFPs), Polyvinyl alcohol, Synthetic polymers | Modify ice crystal structure, inhibit recrystallization, control nucleation | Biomimetic approach; emerging materials show promise for reducing CPA requirements [97] |
| Base Media | Leibovitz L-15, University of Wisconsin solution | Provide ionic and nutrient environment, buffer pH changes | Affect thermal properties; L-15 common for ovarian tissue [98] |
| Additives | Human serum albumin (HSA), Sucrose, Antioxidants | Provide colloidal support, osmotic buffering, reduce oxidative stress | HSA at 4 mg/mL with 0.1M sucrose improves outcomes [98] |
Visualization of Experimental Workflows
Freeze-Thaw Study Design Workflow
Two-Factor Hypothesis Mechanism
Scaling Correlations and Predictive Modeling
Successful scale-translation requires establishing quantitative relationships between small-scale measurements and large-scale outcomes:
Cooling Rate Equivalency: Establish correlation between cooling rates in different container geometries that produce equivalent cell viability outcomes. This often requires accounting for container thermal mass and heat transfer characteristics [55].
CPA Toxicity Scaling: Model time-dependent toxicity effects across different volumes, as toxicity correlates with both CPA concentration and exposure duration. This is particularly critical for DMSO, which demonstrates concentration and time-dependent cytotoxicity [100] [99].
Nucleation Probability Modeling: Account for the volume-dependence of stochastic ice nucleation events, as larger volumes nucleate at higher temperatures on average, affecting ice crystal size distribution [57].
Thermal Gradient Prediction: Develop models that predict the magnitude of thermal gradients in production-scale systems based on measurements in small-scale containers with intentional thermal heterogeneity [55].
Designing small-scale freeze-thaw studies that accurately predict large-scale performance requires a systematic approach grounded in the fundamental biophysics of freezing injury. By implementing the protocols and principles outlined in this technical guide—including comprehensive cooling rate characterization, thermodynamic profiling, and strategic container scale-down—researchers can develop robust correlations between small-scale experiments and manufacturing outcomes. The integration of quantitative modeling with empirical validation creates a powerful framework for protocol optimization and scale-translation, ultimately enhancing the efficiency and success of biopharmaceutical development and cell therapy commercialization.
The future of predictive scale-translation lies in advancing multi-scale modeling approaches that integrate molecular-level interactions between cryoprotectants and biomolecules with container-level heat and mass transfer phenomena. Such integrated models, validated through carefully designed small-scale studies, will continue to improve our ability to design freeze-thaw protocols that maintain consistent cell quality and functionality across all scales of operation.
Conclusion
The precise control of freezing rates is not merely a technical step but a fundamental determinant of success in biopreservation. The central challenge lies in navigating the delicate balance between the two major causes of cell death: intracellular ice formation at high cooling rates and excessive dehydration at slow rates. Emerging strategies, particularly controlled ice nucleation, offer a powerful means to steer this balance, promoting protective dehydration while minimizing destructive ice formation. As the field advances, future directions will focus on developing DMSO-free cryoformulations, creating integrated, non-invasive monitoring systems, and establishing universally predictive, cell-type-specific models. Mastering these principles is paramount for enhancing the efficacy and safety of cell-based therapies, biologics, and the burgeoning field of regenerative medicine, ensuring that these living medicines survive the cold to fulfill their therapeutic potential.
References
- 1. Cooling-rate dependence of the cryopreservation ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224025000434]
- 2. 2. TF-1 cells during graded freezing (interrupted slow ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224010000787]
- 3. Identification of a fundamental cryoinjury mechanism in ... [https://www.sciencedirect.com/science/article/pii/S0011224023000913]
- 4. Impact of controlled ice nucleation on intracellular ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517324009281]
- 5. Protecting Cell Viability During Cryopreservation [https://www.azenta.com/learning-center/blog/considerations-protecting-cell-viability-during-cryopreservation]
- 6. Proteomics and metabolomics analyses of mechanism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11755957/]
- 7. Cryopreservation - Key Survey Findings from the ISCT ... [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/05/28/cryopreservation-key-survey-findings-from-the-isct]
- 8. Role of Cells in Freezing-Induced Cell-Fluid-Matrix ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3708713/]
- 9. Optimizing cryopreservation strategies for scalable cell therapies [https://pmc.ncbi.nlm.nih.gov/articles/PMC11659802/]
- 10. Cryopreservation: An Overview of Principles and Cell- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7995302/]
- 11. Measurement of Water Transport during Freezing in Cell ... [https://www.sciencedirect.com/science/article/pii/S001122409792071X]
- 12. The Impact of Varying Cooling and Thawing Rates on ... [https://www.nature.com/articles/s41598-019-39957-x]
- 13. Effects of Freezing, Frozen Storage and Thawing on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12331523/]
- 14. Effect of freezing rate on water state in strawberry [https://www.sciencedirect.com/science/article/abs/pii/S0308814625036593]
- 15. Relevance of controlled cooling and freezing phases in T ... [https://pubmed.ncbi.nlm.nih.gov/38768801/]
- 16. The transfer temperature from slow cooling to cryogenic storage ... [https://ncbi.nlm.nih.gov/pmc/articles/PMC8594829]
- 17. An Improved Model for Nucleation-Limited Ice Formation in Living Cells during Freezing | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0098132]
- 18. Kinetics of Intracellular Ice Formation in One-Dimensional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1305042/]
- 19. A Low Temperature Limit for Life on Earth | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0066207]
- 20. The impact of initial cooling rates on cell preservation in frozen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12408769/]
- 21. Ice formation and its elimination in cryopreservation of ... [https://www.nature.com/articles/s41598-024-69528-8]
- 22. Targeting Ice Recrystallization to Improve Cryopreservation ... [https://www.genengnews.com/topics/translational-medicine/targeting-ice-recrystallization-to-improve-cryopreservation-consistency/]
- 23. CN112292033A - 改进的超快冷却系统和使用方法 [https://patents.google.com/patent/CN112292033A/zh]
- 24. Cryopreservation in Cell and Gene Therapy [https://cgt.global/cryopreservation-in-cell-and-gene-therapy-ensuring-high-viability-cells-for-research-manufacturing/]
- 25. Cryopreservation of NK and T cells without DMSO for adoptive ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376086/]
- 26. A Network Model of Intracellular Ice Formation | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0058343]
- 27. Impact of controlled ice nucleation on intracellular ... [https://pubmed.ncbi.nlm.nih.gov/39265855/]
- 28. Sulfoxide-functional trehalose enhances DMSO-free ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894724051957]
- 29. Control of stress-induced apoptosis by freezing tolerance ... [https://www.sciencedirect.com/science/article/pii/S1355814523015560]
- 30. Insights on the role of cryoprotectants in enhancing the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11729100/]
- 31. Classical nucleation theory [https://en.wikipedia.org/wiki/Classical_nucleation_theory]
- 32. Brief Overview of Ice Nucleation - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7828621/]
- 33. Chemical approaches to cryopreservation [https://www.nature.com/articles/s41570-022-00407-4]
- 34. Impact of Freezing and Freeze Drying on Lactobacillus ... [https://www.mdpi.com/2304-8158/14/10/1817]
- 35. Overcoming ice: cutting-edge materials and advanced ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11887326/]
- 36. Freezing vs. vitrification [https://bio-protocol.org/exchange/minidetail?id=9216502&type=30]
- 37. A review of best practices of rapid-cooling vitrification for ... [https://www.asrm.org/practice-guidance/practice-committee-documents/a-review-of-best-practices-of-rapid-cooling-vitrication-for-oocytes-and-embryos-a-committee-opinion-2021/]
- 38. Comparison of the quality of ovarian tissue cryopreservation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11948982/]
- 39. Comparative effectiveness of vitrification and slow freezing ... [https://bmcwomenshealth.biomedcentral.com/articles/10.1186/s12905-024-03505-1]
- 40. The impact of initial cooling rates on cell preservation in ... [https://link.springer.com/article/10.1007/s44211-025-00815-8]
- 41. Cell Storage Protocols: Best Practices for Cell Survival [https://www.corning.com/worldwide/en/products/life-sciences/resources/stories/at-the-bench/cell-storage-protocols-best-practices-for-cell-survival.html]
- 42. Advanced Cryopreservation Techniques for Long-Term ... [https://www.cytion.com/us/Knowledge-Hub/Blog/Advanced-Cryopreservation-Techniques-for-Long-Term-Cell-Storage/]
- 43. Effect of different freezing rates during cryopreservation of rat ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3465236/]
- 44. Walking on thin ice: controlled freezing & thawing of ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/2677/Walking-on-thin-ice-controlled-freezing-thawing-of-pharmaceutical-active-molec]
- 45. [1312.0822] Homogeneous at moderate ice ... nucleation supercooling [https://arxiv.org/abs/1312.0822]
- 46. Prior heterogeneous ice nucleation events shape ... - ACP [https://acp.copernicus.org/articles/25/13995/2025/]
- 47. The Importance of Controlling the Rate of Freezing [https://www.thermofisher.com/blog/life-in-the-lab/the-importance-of-controlling-the-rate-of-freezing/]
- 48. Impact of Cryopreservation and Freeze-Thawing on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9045030/]
- 49. Comparison of ice fog methods and monitoring of controlled ... [https://pubmed.ncbi.nlm.nih.gov/30597272/]
- 50. Manipulating Ice Nucleation and Growth During Freeze-Casting [https://research.manchester.ac.uk/en/studentTheses/manipulating-ice-nucleation-and-growth-during-freeze-casting/]
- 51. Controlled Ice Using ControLyo... | SpringerLink Nucleation [https://link.springer.com/protocol/10.1007/978-1-4939-8928-7_3]
- 52. Principles and biotechnological applications of bacterial ice nucleation [https://pubmed.ncbi.nlm.nih.gov/1760850/]
- 53. Microphysical detection of nano-ice nuclei to ice crystals [https://www.nature.com/articles/s41612-025-01062-4]
- 54. ACP - HUB: a method to model and extract the distribution of ice ... [https://acp.copernicus.org/articles/23/5623/2023/]
- 55. Modeling and typical cases analyze at the cell-scale of ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224025000161]
- 56. Improving Cell Recovery: Freezing and Thawing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8909336/]
- 57. Damage due to ice crystallization | Scientific Reports [https://www.nature.com/articles/s41598-025-86117-5]
- 58. The effects of cryopreservation on PBMCs transcriptome ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1690316/full]
- 59. Understanding the freezing responses of T-cells and other ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7682753/]
- 60. Mechanisms of freezing damage [https://pubmed.ncbi.nlm.nih.gov/3332492/]
- 61. Freezing of living cells: mechanisms and implications [https://pubmed.ncbi.nlm.nih.gov/6383068/]
- 62. Effects of freezing on membranes and proteins in LNCaP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1994664/]
- 63. Protective effect of intracellular ice during freezing? [https://pubmed.ncbi.nlm.nih.gov/12686211/]
- 64. New insights into the mechanism of freeze-induced ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996922008158]
- 65. Applications and optimization of cryopreservation ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/415/Applications-and-optimization-of-cryopreservation-technologies-to-cellular-therapeutics]
- 66. Comparison of the effects of three cryoprotectants on ... [https://www.spandidos-publications.com/10.3892/etm.2020.9076]
- 67. Optimisation of cryopreservation conditions, including storage ... [https://bmcmolcellbiol.biomedcentral.com/articles/10.1186/s12860-024-00516-6]
- 68. The Temperature and Type of Intracellular Ice Formation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2874502/]
- 69. Interrupted cooling protocols in cryopreservation: A review ... [https://pubmed.ncbi.nlm.nih.gov/41192344/]
- 70. Intracellular ice formation is affected by cell interactions [https://pubmed.ncbi.nlm.nih.gov/10413578/]
- 71. The Impact of Annealing Methods on the Encapsulating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11362187/]
- 72. The influence of magnetic field-assisted osmotic ... [https://www.sciencedirect.com/science/article/abs/pii/S0140700725004153]
- 73. Bridging polymer chemistry and cryobiology [https://www.nature.com/articles/s41428-022-00735-8]
- 74. How to Reduce Cryo-concentration With World-class Tools in Freeze/thaw Process [https://blog.entegris.com/how-to-reduce-cryo-concentration-with-world-class-tools-in-freeze/thaw-process]
- 75. Freeze–thaw characterization process to minimize ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8166975/]
- 76. Advanced technologies for the preservation of mammalian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8765766/]
- 77. From cold chain to ambient: Benefits, risks, and evidence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12593556/]
- 78. Evaluation of the viability and functionality of human ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1627973/full]
- 79. Towards a universal rapid warming protocol for ... [https://www.sciencedirect.com/science/article/pii/S147264832500358X]
- 80. Probing mechanisms of cryopreservation damage to ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925000350]
- 81. Characterization of Water States in Canola Seeds With ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12621169/]
- 82. Low-field NMR-based deep learning for non-destructive ... [https://www.sciencedirect.com/science/article/abs/pii/S030881462502432X]
- 83. 1H NMR Relaxation Processes in Lung Tissues at Low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525575/]
- 84. Dose-efficient cryo-electron microscopy for thick samples ... [https://www.nature.com/articles/s41592-025-02834-9]
- 85. Cryo-imaging gives deeper view of thick biological materials [https://news.cornell.edu/stories/2025/10/cryo-imaging-gives-deeper-view-thick-biological-materials]
- 86. Cryo-EM and Solid State NMR Together Provide a ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/38853912/]
- 87. Walking on thin ice: controlled freezing & thawing of ... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/2677/Walking-on-thin-ice-controlled-freezing-thawing-of-pharmaceutical-active-molecules]
- 88. Cryopreservation and post-thaw differentiation of monocytes ... [https://pubs.rsc.org/en/content/articlehtml/2025/lp/d5lp00131e]
- 89. Viable and Functional: Long-Term −80 °C Cryopreservation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12525932/]
- 90. Impact of pre- and post-thaw processing on recovery ... [https://www.sciencedirect.com/science/article/abs/pii/S1465324925008400]
- 91. Cryopreservation of biological materials: applications and ... [https://link.springer.com/article/10.1007/s11626-025-01027-0]
- 92. Reduced Pressure Ice Fog Technique for Controlled ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2799604/]
- 93. Cell Freezing Media Market to Grow 8.6% CAGR till 2035 [https://www.towardshealthcare.com/insights/cell-freezing-media-market-sizing]
- 94. Cell Freezing Media Market | Global Market Analysis Report [https://www.futuremarketinsights.com/reports/cell-freezing-media-market]
- 95. Rapid freezing versus slow programmable ... [https://www.sciencedirect.com/science/article/pii/S0015028208048206]
- 96. Comparison between Slow Freezing and Vitrification in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10374242/]
- 97. Overcoming ice: cutting-edge materials and advanced ... [https://jnanobiotechnology.biomedcentral.com/articles/10.1186/s12951-025-03265-6]
- 98. Optimization of freezing and thawing protocols for human ... [https://www.sciencedirect.com/science/article/pii/S0011224025000513]
- 99. Impact of Freeze–Thaw Processes on the Quality of Cells [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/365/Impact-of-FreezeThaw-Processes-on-the-Quality-of-Cells]
- 100. Cell freezing and the biology of inexorability - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11564080/]