Cellular Cryobiology: Unraveling the Mechanical and Osmotic Effects of Freezing on Cell Integrity and Preservation
This article provides a comprehensive examination of the mechanical and osmotic stresses that cells encounter during freezing and thawing processes, crucial for researchers and professionals in drug development and biopreservation.
Cellular Cryobiology: Unraveling the Mechanical and Osmotic Effects of Freezing on Cell Integrity and Preservation
Abstract
This article provides a comprehensive examination of the mechanical and osmotic stresses that cells encounter during freezing and thawing processes, crucial for researchers and professionals in drug development and biopreservation. It explores the fundamental biophysical mechanisms of freezing-induced cellular damage, including intracellular ice formation, membrane phase transitions, and dehydration-driven mechanical stress. The content details advanced preservation methodologies such as slow freezing and vitrification, alongside optimization strategies involving cryoprotectant agents and controlled rate technologies. By synthesizing foundational theories with current research and validation approaches, this resource aims to enhance survival rates and functional integrity of preserved cells for therapeutic and research applications.
The Biophysics of Freezing Injury: Understanding Osmotic Stress and Mechanical Damage in Cells
Fundamental Mechanisms of Freezing-Induced Cell Damage
The preservation of living cells at ultra-low temperatures is a cornerstone of modern biotechnology, medical research, and clinical applications. Despite its widespread use, cryopreservation remains a significant bottleneck in the cell therapy industry, often introducing variability and compromising cellular attributes [1]. The fundamental challenge lies in navigating the two predominant, and often competing, physical mechanisms of freezing-induced cell damage: mechanical damage from intracellular ice formation and osmotic stress from solute concentration and cell dehydration [2] [3]. Understanding the delicate balance between these pathways is crucial for developing robust preservation protocols for sensitive cell types, including stem cells, natural killer (NK) cells, and mesenchymal stem cells (MSCs), which are vital for regenerative medicine and cancer immunotherapy [4] [5] [1]. This whitepaper provides an in-depth analysis of the core mechanisms of cryoinjury, supported by quantitative data and experimental methodologies, to inform researchers and drug development professionals in their pursuit of optimized cell preservation strategies.
Fundamental Mechanisms of Cryoinjury
Mechanical Damage: Intracellular Ice Formation
Intracellular ice formation (IIF) is widely considered the most lethal event during freezing. It occurs when the cooling rate is too rapid to permit sufficient cellular dehydration, resulting in the supercooling of intracellular water until it nucleates and forms ice crystals [2]. These crystals mechanically disrupt organelles, the cytoskeleton, and the plasma membrane, leading to immediate and irreversible cell death [3]. The likelihood of IIF is a direct function of the cooling rate. Mazur's classic "two-factor hypothesis" established the kinetic basis for this phenomenon, describing how water transport across the cell membrane is outpaced by cooling, leaving water trapped inside the cell to freeze [2]. The cell's membrane permeability to water and its surface area-to-volume ratio are critical determinants of its susceptibility to IIF. Consequently, cell types with large surface area-to-volume ratios, such as oocytes, are particularly vulnerable [5].
Osmotic Damage: Solute Effects and Cell Dehydration
The slow freezing of cells initiates a sequence of osmotic imbalances that constitute the second major pathway of cryoinjury. As ice forms in the extracellular solution, solutes are excluded from the growing ice lattice, leading to a progressive concentration of electrolytes and other solutes in the remaining unfrozen medium [2]. This creates a hypertonic environment, causing water to osmotically exit the cell. The cell shrinks and undergoes substantial dehydration. Historically, this damage was attributed to the concentrated solutes themselves, which can denature proteins and disrupt lipid bilayers. However, more recent evidence suggests that the physical reduction in the size of the unfrozen channels surrounding the cell, and the associated cell shrinkage, are more injurious [2]. Furthermore, during thawing, cells can experience osmotic shock if the cryoprotectant is not diluted properly, as water rushes into the shrunken cells too rapidly, potentially causing lysis [5].
Table 1: Primary Mechanisms of Freezing-Induced Cell Damage
| Damage Mechanism | Primary Cause | Consequence on Cell | Key Cell Types Affected |
|---|---|---|---|
| Intracellular Ice Formation | Excessively rapid cooling rate | Mechanical rupture of membranes and organelles; immediate cell death | Oocytes [5], human iPSC [5] |
| Cell Dehydration | Slow cooling; extracellular ice formation | Critical volume reduction; solute concentration; membrane damage | Natural Killer (NK) cells [4], MSCs [1] |
| Solution Effects | Concentration of solutes in unfrozen fraction | Protein denaturation; membrane disruption | Broadly affects all cell types [2] |
| Cryoprotectant Toxicity | Chemical effects of CPAs (e.g., DMSO) | Altered membrane fluidity; reduced cytotoxicity (in NK cells) [4] | NK cells [4], various stem cells [3] |
Quantitative Assessment of Cryopreservation Outcomes
A systematic, quantitative approach is essential for evaluating the impact of cryopreservation on different cell types. The following data, compiled from recent studies, highlights the variable responses of cells to freezing and thawing.
Table 2: Quantitative Impact of Cryopreservation on Cell Viability and Function
| Cell Type | Post-Thaw Viability/Recovery | Impact on Function | Optimal Cooling Rate | Key Findings |
|---|---|---|---|---|
| Natural Killer (NK-92) | Not specified | Reduced cytotoxicity & membrane fluidity after CPA exposure [4] | 4-5 °C/min [4] | Damage linked to disrupted cytolytic granules (perforin, granzyme) [4] |
| Induced Pluripotent Stem Cells (iPSC) | Recovery in 4-7 days (optimized); up to 2-3 weeks (unoptimized) [5] | Not specified | -1 °C/min to -3 °C/min [5] | High vulnerability to intracellular ice; growth phase before freezing is critical [5] |
| Bone Marrow-MSCs | Viability reduced at 0h, recovers by 24h [1] | Metabolic activity & adhesion reduced for >24h; variable differentiation potential [1] | -1 °C/min [1] | Fresh and cryopreserved MSCs are functionally different [1] |
| Lactobacillus rhamnosus GG | 90.94% (fast freezing in LN₂); 2% (suboptimal freeze-drying) [6] | Not specified | Not specified | Fast freezing in LN₂ resulted in highest survival [6] |
Experimental Protocols for Investigating Cryoinjury
Protocol: Controlled-Rate Freezing and Thawing of Human Cells
This standard protocol is adapted for human BM-MSCs and iPSCs and is fundamental for investigating osmotic and mechanical damage [5] [1].
Materials:
- Log-phase cells at high viability (>90%)
- Freezing Medium: Culture medium supplemented with 10-20% FBS and 10% DMSO, or a commercial serum-free alternative like Synth-a-Freeze [7].
- Cryogenic vials
- Controlled-rate freezing apparatus (e.g., "Mr. Frosty" or programmable freezer)
- Liquid nitrogen storage tank
- Water bath (37°C-40°C)
Method:
- Harvesting: Detach adherent cells gently using a dissociation reagent like trypsin. Quench the reaction with complete growth medium [7].
- Preparation: Centrifuge the cell suspension (100-400 x g for 5-10 min), aspirate the supernatant, and resuspend the pellet in cold freezing medium at a concentration of 1-5 x 10⁶ cells/mL [1] [7].
- Aliquoting: Dispense 1 mL of cell suspension into each cryovial. Mix the suspension often to ensure homogeneity during aliquoting.
- Freezing: Place the cryovials in a controlled-rate freezing apparatus. Cool the cells at approximately -1°C/min to -80°C. For many cell types, this slow cooling is critical to allow for sufficient dehydration and avoid IIF [5] [1] [7].
- Storage: After 24 hours, transfer the vials to the vapor phase of a liquid nitrogen tank for long-term storage (< -135°C) to prevent stressful thermal transitions above the glass transition temperature [5] [7].
- Thawing: Rapidly thaw the cells by gently agitating the vial in a 37°C water bath for about 1 minute until only a small ice crystal remains [1].
- Dilution & Washing: Transfer the cell suspension to a tube containing 9-10 mL of pre-warmed culture medium. This stepwise dilution is critical to prevent osmotic shock. Centrifuge to remove the DMSO-containing medium and resuspend the cell pellet in fresh culture medium for subsequent assays [5] [1].
Protocol: Assessing Membrane Integrity and Apoptosis Post-Thaw
Quantifying viability and apoptosis at multiple time points post-thaw is crucial for a complete picture of cryoinjury, as cell death can be delayed [1].
Materials:
- Post-thaw cell suspension
- Trypan Blue stain or automated cell counter (e.g., Countess)
- Flow cytometer
- Annexin V / Propidium Iodide (PI) apoptosis detection kit
- Phosphate Buffered Saline (PBS)
Method:
- Immediate Viability: At 0 hours post-thaw, mix a cell sample with Trypan Blue and count viable (unstained) and non-viable (blue) cells using a hemocytometer or automated counter [1] [7].
- Delayed Apoptosis Assessment: At 0h, 2h, 4h, and 24h post-thaw, collect and wash cells with PBS.
- Staining: Resuspend cells in Annexin V binding buffer and stain with Annexin V-FITC and PI according to the manufacturer's protocol. Use unstained and single-stained controls for compensation.
- Flow Cytometry: Analyze the samples on a flow cytometer. Distinguish live cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic/necrotic cells (Annexin V+/PI+), and cells that have died through primary necrosis (Annexin V-/PI+).
- Analysis: The percentage of cells in early apoptosis typically increases over the first 4 hours post-thaw before dropping by 24 hours as viability stabilizes, indicating a recovery period is needed for the population [1].
Diagram 1: Two primary pathways of freezing-induced cell damage. Slow cooling primarily causes osmotic damage from dehydration, while rapid cooling causes mechanical damage from intracellular ice.
Advanced and Emerging Research
Novel Cryoprotective Strategies
Research is actively moving beyond traditional CPAs like DMSO to develop safer and more effective materials. A promising approach involves the use of membrane-targeted DNA frameworks (DFs) [8]. These nanoscale structures, functionalized with cholesterol (Chol24-DF), are engineered to specifically anchor to the cell membrane. Unlike DMSO, which acts colligatively, Chol24-DF provides a physical scaffold that stabilizes the membrane against the mechanical stresses of freezing and inhibits ice recrystallization. A key advantage is its biodegradability; the DNA structure degrades under physiological conditions post-thaw, eliminating long-term toxicity concerns associated with DMSO retention [8].
Another strategy involves using combinations of permeating and non-permeating agents to create vitrification mixtures. This allows for a reduction in the concentration of toxic PAs like DMSO while maintaining cryoprotective efficacy. For example, sugars like trehalose and sucrose are effective NPAs. Trehalose, with its stable α-1,1-glycosidic bond, is particularly effective at stabilizing membranes and proteins in a dry state, mimicking the natural protectants found in stress-tolerant organisms [3]. The addition of osmolytes to CPA cocktails has also been shown to mitigate the loss of membrane fluidity and cytotoxicity in NK cells exposed to cryoprotectants before freezing [4].
Cell-Specific Considerations and the Recovery Period
The optimal cryopreservation protocol is highly cell type-specific. For instance, while a cooling rate of -1°C/min is standard for MSCs and hematopoietic stem cells, oocytes, pancreatic islets, and embryonic stem cells often benefit from more rapid cooling [3]. Furthermore, the physical state of the cells during freezing (e.g., as single cells or aggregates) impacts recovery. Freezing iPSCs as aggregates can preserve cell-cell contacts that support survival, but it can also create variability in cryoprotectant penetration [5].
A critical, often overlooked, factor is the post-thaw recovery period. Quantitative studies on BM-MSCs show that while cell viability may recover within 24 hours, functional attributes like metabolic activity and adhesion potential can remain impaired for longer [1]. This implies that a 24-hour period is insufficient for a full functional recovery, which has significant implications for clinical applications where cells are infused shortly after thawing.
Diagram 2: General workflow for a controlled-rate freezing and post-thaw analysis experiment.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Cryopreservation Studies
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating Cryoprotectant Agent (CPA) | Standard 10% (v/v) in culture medium for most mammalian cells [7] [3]. |
| Synth-a-Freeze Medium | Chemically defined, protein-free freezing medium | Cryopreservation of stem and primary cells without animal serum [7]. |
| Trehalose | Non-permeating CPA; stabilizes membranes and proteins | Added to CPA cocktails to improve survival during freeze-drying of probiotics [6] and vitrification mixtures [3]. |
| Annexin V / PI Apoptosis Kit | Flow cytometry-based detection of apoptosis and necrosis | Quantifying delayed-onset apoptosis in MSCs at 0h, 2h, 4h, and 24h post-thaw [1]. |
| Controlled-Rate Freezer (e.g., "Mr. Frosty") | Apparatus to achieve a consistent, slow cooling rate (~ -1°C/min) | Standard freezing protocol for iPSCs and MSCs to avoid intracellular ice formation [5] [1]. |
| DNA Framework (Chol24-DF) | Emerging membrane-targeted cryoprotectant | Investigated for protecting macrophage cell lines (RAW264.7) with enhanced membrane targeting and biodegradability [8]. |
| Skim Milk Powder | Protective agent forming a rigid matrix to inhibit ice crystal growth | Used as a stabilizer in combination with trehalose for freeze-drying probiotics like LGG [6]. |
Intracellular ice formation (IIF) is widely recognized as a lethal event in cryobiology, playing a critical role in cell destruction during cryosurgery and presenting a major obstacle for successful cryopreservation [9]. When cells are cooled rapidly, the internal water content does not have sufficient time to permeate out through the membrane via osmotic efflux, leading to supercooled conditions that favor the nucleation of ice crystals within the cytoplasm [10]. This phenomenon represents a crucial aspect of the broader mechanical and osmotic effects of freezing on cellular systems, with IIF serving as a primary mechanism of direct cell injury [9].
The lethal nature of intracellular ice stems from the mechanical damage inflicted upon essential cellular structures. The growth of ice crystals within the confined intracellular space can disrupt organelles, perforate membrane systems, and compromise structural integrity, leading to irreversible cellular damage [9]. Understanding the precise conditions and mechanisms governing IIF is therefore fundamental to advancing both destructive applications like cryosurgery and protective applications like cell preservation.
Mechanisms of Intracellular Ice Formation
Physical Processes and Pathways
The formation of intracellular ice involves a complex interplay of physical processes during cooling. Two primary mechanisms have been proposed for how ice enters the cell: pore nucleation and membrane damage.
Pore Nucleation Theory: This hypothesis suggests that the plasma membrane contains aqueous pores that can serve as conduits for ice propagation into the intracellular space. According to this view, extracellular ice crystals can initiate the freezing of intracellular water through these membrane pores [10].
Membrane Damage Hypothesis: Experimental evidence from cultured mouse fibroblasts challenges the pore nucleation theory, indicating instead that the plasma membrane may be damaged at a critical gradient in osmotic pressure across the membrane. This damage then facilitates the nucleation of intracellular ice [10].
The cooling rate profoundly influences which mechanism dominates. At slow cooling rates (typically <10°C/min), cells have sufficient time to dehydrate in response to extracellular ice formation, reducing the likelihood of IIF. In contrast, rapid cooling (>50°C/min) traps water inside the cell, creating conditions favorable for intracellular nucleation [11].
Molecular Interactions and Nucleation
At the molecular level, ice nucleation represents a complex process involving the organization of water molecules into crystalline structures. Heterogeneous nucleation—where foreign surfaces or particles catalyze ice formation—plays a crucial role in IIF. Kaolinite, a clay mineral, demonstrates this effect by enhancing ice nucleation rates by approximately 20 orders of magnitude compared to homogeneous nucleation in pure water [12].
Classical nucleation theory provides a framework for understanding IIF, though experimental observations reveal deviations from ideal behavior. Critical nuclei for intracellular ice display a strong two-dimensional character rather than the spherical caps predicted by theory, particularly when forming at interfaces [12]. This anisotropic growth pattern reflects the influence of cellular structures and membranes on the crystallization process.
Table 1: Key Factors Influencing Intracellular Ice Formation
| Factor | Effect on IIF | Experimental Evidence |
|---|---|---|
| Cooling Rate | High cooling rates (>50°C/min) promote IIF by limiting cellular dehydration | Mouse oocyte studies showing increased IIF with rapid cooling [13] |
| Extracellular Ice | Serves as potential nucleation source through membrane interactions | Cryomicroscopy of fibroblasts showing correlation between extracellular ice and IIF [10] |
| Membrane Properties | Hydraulic conductivity and surface area regulation affect water efflux | Modeling of mouse oocytes accounting for membrane transport [13] |
| Solution Effects | High solute concentration depresses freezing point but increases osmotic stress | Mazur's two-factor hypothesis balancing IIF and solute effects [11] |
Experimental Evidence and Methodologies
Critical Experimental Findings
Seminal investigations using cryomicroscopy have provided direct visual evidence of intracellular ice formation and its lethal consequences. In designed experiments with cultured mouse fibroblasts, researchers critically assessed prevailing hypotheses about IIF genesis. The experimental data did not support theories involving critical undercooling, aqueous pore nucleation, or electrical transients at the ice interface. Instead, evidence pointed toward membrane damage at critical osmotic pressure gradients as the initiating event for IIF [10].
In cryosurgical applications, experiments in vivo have demonstrated that intracellular ice formation contributes significantly to direct cell injury. The sequence of events begins with ice crystal formation which removes water from cells and initiates a cascade of deleterious events [9]. Recent investigations have identified that cell death occurs through necrosis in the central part of cryogenic lesions where IIF is most extensive, while apoptosis predominates in peripheral zones with less severe freezing [9].
Quantitative Methodologies and Modeling
Advanced modeling approaches have been developed to predict intracellular ice formation and its consequences. A recent cell-scale model incorporates transmembrane transport of water and cryoprotectants alongside intracellular crystallization and recrystallization during the freeze-thaw process [13]. This comprehensive model represents a significant advancement as it describes recrystallization during rewarming—a previously neglected but critical aspect of IIF damage.
The experimental determination of IIF kinetics employs sophisticated techniques:
- Cryomicroscopy with image analysis: Quantifying "blackening" or darkening of cells as ice forms
- Forward flux sampling (FFS): Computing nucleation rates at the molecular level using atomistic models
- Differential scanning calorimetry (DSC): Measuring ice crystallization thermodynamics
- Splat assays: Visualizing ice crystal size and structure under polarized light
These methodologies have revealed that the critical nucleus size for intracellular ice is substantially smaller in heterogeneous nucleation (approximately 225 water molecules) compared to homogeneous nucleation (approximately 540 water molecules) [12].
Table 2: Experimental Models for Studying Intracellular Ice Formation
| Experimental Model | Key Applications | Technical Advantages |
|---|---|---|
| Mouse fibroblasts (in vitro) | Testing IIF hypotheses, membrane damage studies | Controlled environment, direct observation via cryomicroscopy [10] |
| Mouse oocytes | Kinetics of IIF and recrystallization | Large cell size facilitates observation, standardized freezing protocols [13] [14] |
| Transplanted tumors (in vivo) | Cryosurgical mechanisms, apoptosis/necrosis balance | Physiological relevance, vascular effects [9] |
| Computational models | Predicting IIF under various conditions | Non-invasive parameter testing, molecular-level insights [13] [12] |
Quantitative Analysis of Intracellular Ice
The formation and growth of intracellular ice follow quantifiable kinetic patterns that can be mathematically modeled. For mouse oocytes subjected to interrupted rapid cooling, recrystallization of intracellular ice follows temperature-dependent kinetics with measurable activation energy [14]. The blackening score—a visual indicator of ice formation—increases with time at holding temperatures between -65°C and -50°C, with the highest rates observed at -50°C [14].
The cooling rate dramatically affects both the probability of IIF and the resulting cell survival. Mazur's two-factor hypothesis establishes that optimal cooling rates must balance the risk of intracellular ice formation (favored by rapid cooling) against osmotic injury and solute effects (favored by slow cooling) [11]. This balance varies significantly between cell types due to differences in membrane permeability and surface area regulation.
Consequences and Implications
Cellular Damage Mechanisms
Intracellular ice formation inflicts damage through multiple mechanisms that collectively ensure cell destruction:
- Mechanical Damage: Physical disruption of organelles and membrane systems by growing ice crystals [9]
- Osmotic Imbalance: Alteration of solute concentrations leading to volume dysregulation [15]
- Oxidative Stress: Generation of reactive oxygen species during freezing and thawing [15]
In cryosurgical applications, these direct cellular injuries synergize with vascular injury mechanisms. After thawing, the microcirculation in previously frozen tissue progressively fails, resulting in vascular stasis within approximately one hour. This circulatory failure ensures comprehensive cell death through ischemia, particularly in peripheral zones where IIF alone may be insufficient to destroy all cells [9].
Implications for Cryopreservation and Cryosurgery
The control of intracellular ice formation has profound implications for both protective and destructive freezing applications:
Cryopreservation Strategies: Successful cell preservation requires protocols that minimize IIF through optimized cooling rates and cryoprotective agents. Mathematical models that predict IIF probability enable the design of freezing protocols that avoid damaging gradients in osmotic pressure [10] [13]
Cryosurgical Efficacy: In contrast, cryosurgical techniques aim to maximize tissue destruction through controlled induction of IIF. The strategic application of rapid cooling promotes lethal intracellular ice formation in targeted tissues [9]
Novel Cryoprotectants: Recent research has identified compounds like tricine that demonstrate multiple protective functions, including osmotic regulation, ice recrystallization inhibition, and antioxidant activity. Such multi-functional agents show promise for improving cryopreservation outcomes while minimizing IIF [15]
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials for Intracellular Ice Studies
| Reagent/Material | Function/Application | Experimental Notes |
|---|---|---|
| Cryomicroscope | Direct visualization of ice formation in cells | Custom-built stages allow precise temperature control [10] |
| Dimethyl Sulfoxide (DMSO) | Penetrating cryoprotectant | 5-10% concentrations common; affects membrane properties [11] [16] |
| Glycerol | Penetrating cryoprotectant | 20-40% for RBC cryopreservation; requires deglycerolization [11] [15] |
| Tricine | Multi-functional cryoprotectant | Osmotic regulation, IRI, and antioxidant properties [15] |
| Hydroxyethyl Starch (HES) | Non-penetrating cryoprotectant | Stabilizes cell membrane; inhibits ice formation [15] |
| Mouse Oocytes | Model system for IIF kinetics | Large size facilitates observation; well-characterized [13] [14] |
| Kaolinite | Heterogeneous ice nucleator | Boosts ice nucleation by 20 orders of magnitude [12] |
| Differential Scanning Calorimeter | Quantification of ice crystallization | Measures bound water ratio and crystallization thermodynamics [15] |
Intracellular ice formation represents a primary lethal mechanism during rapid cooling of biological systems, with significant implications across cryobiology, from cryopreservation to cryosurgery. The physical processes governing IIF involve complex interactions between cooling rate, membrane properties, and nucleation phenomena, which can be quantitatively analyzed through advanced experimental and computational approaches.
Current research continues to refine our understanding of IIF mechanisms, with recent investigations highlighting the importance of recrystallization during warming and the potential of novel cryoprotectants that target multiple damage pathways. These advances support the development of more effective strategies for both cell preservation and destruction, framed within the broader context of mechanical and osmotic effects of freezing on cellular systems.
Osmotic Dehydration and Solute Effects During Slow Freezing
Slow freezing is a fundamental technique in cryopreservation, where the controlled reduction of temperature induces complex osmotic and mechanical responses in biological systems. This process initiates when extracellular water begins to freeze, increasing the concentration of solutes in the remaining unfrozen fluid. This creates a pronounced osmotic gradient across cell membranes, driving intracellular water out of cells and leading to cellular dehydration [17]. The rate of cooling critically determines the extent and effects of this dehydration. Optimal slow freezing rates facilitate sufficient water efflux to avoid lethal intracellular ice formation, yet excessive dehydration can concentrate intracellular solutes to toxic levels and cause damaging cell shrinkage [18].
Understanding these osmotic and solute effects is paramount for preserving cellular viability and function across diverse fields. In drug development, cryopreservation ensures the stability of cellular therapeutic products like CAR-T cells, where post-thaw viability directly impacts treatment efficacy [19]. In tissue engineering, successful preservation of bioartificial tissues requires maintaining both cell viability and the mechanical integrity of the extracellular matrix, which can be compromised by freezing-induced fluid redistribution and structural deformation [20] [17]. Similarly, in the food industry, controlling osmotic dehydration during freezing processes like dehydrofreezing helps preserve the texture, nutritional content, and quality of delicate tissues such as fruits [21].
This technical guide synthesizes current research to provide a comprehensive resource on the biophysical principles, experimental data, and methodological protocols underlying osmotic dehydration during slow freezing, with the aim of empowering researchers to optimize cryopreservation outcomes in their specific applications.
Core Principles and Biophysical Mechanisms
The Osmotic Crisis During Freezing
The phase change of water from liquid to ice is the primary driver of osmotic dehydration during slow freezing. As extracellular ice forms, dissolved solutes (salts, sugars, CPAs) are excluded from the crystal lattice, leading to their progressive concentration in the diminishing volume of unfrozen liquid. This phenomenon, known as freeze concentration, dramatically elevates the osmolarity of the extracellular environment [22].
Cells respond to this escalating osmotic stress as predicted by the Boyle-Mariotte relation. Water rapidly exits the cell along its chemical potential gradient, moving from the hypotonic intracellular space to the hypertonic, unfrozen extracellular fluid. This efflux of water causes the cell to shrink and its internal contents to become concentrated. The extent of this dehydration is governed by the cooling rate:
- Slow Cooling: Allows sufficient time for water to leave the cell, minimizing the risk of intracellular ice formation but potentially leading to excessive "solution effects" injury from high solute concentrations and extreme cell volume reduction.
- Rapid Cooling: Outpaces the cell's ability to dehydrate, increasing the probability of water supercooling and eventually forming intracellular ice, which is almost always lethal [18].
The cell membrane acts as a semi-permeable barrier, and its hydraulic permeability (Lp) to water is a critical parameter determining the cell's dehydration kinetics. This permeability is highly temperature-dependent, typically decreasing as temperatures fall, which can further complicate the dehydration process.
Solute Effects and Cryoprotection
The specific solutes present in the system profoundly influence the cellular response to freezing. Their effects can be categorized as damaging or protective.
- Electrolytes: The concentration of electrolytes like sodium and potassium chloride during freeze concentration can disrupt lipid membranes and denature proteins. This "solute effect" is a major cause of cell injury during slow freezing.
- Cryoprotective Agents (CPAs): CPAs are employed to mitigate freezing damage. They are broadly classified as:
- Permeating CPAs: Small, neutral molecules like Dimethyl Sulfoxide (DMSO) and glycerol can cross the cell membrane. They reduce the fraction of intracellular water that can freeze and dampen the increase in intracellular electrolyte concentration, thereby protecting against both intracellular ice formation and solute damage [19] [18].
- Non-Permeating CPAs: Larger molecules or those that do not cross the membrane, such as sucrose, trehalose, and certain synthetic zwitterions, act colligatively by increasing the osmolarity of the extracellular solution. This promotes more gentle, controlled cellular dehydration before the onset of major extracellular freezing. They also help stabilize membrane proteins and cell membranes [19] [18] [22].
Emerging research highlights the role of zwitterions—molecules possessing both positive and negative charges. A study on lipid nanoparticles (LNPs) demonstrated that the zwitterion betaine can be incorporated into particles during freeze-thaw via freeze concentration. Once inside, betaine acts as a proton sponge in the acidic environment of endosomes, enhancing the escape and delivery efficacy of mRNA therapeutics, showcasing a functional benefit beyond mere stabilization [22].
Intercellular and Tissue-Level Effects
In multicellular systems like tissues and spheroids, osmotic effects are compounded by physical connections and tissue biomechanics.
- Gap Junction Communication: In cell spheroids, the formation of intracellular ice in one cell can propagate to adjacent cells through gap junctions, making the cryopreservation of multicellular systems more challenging than single cells [18].
- Freezing-Induced Tissue Deformation: Extracellular ice formation causes a spatiotemporal redistribution of interstitial fluid, leading to swelling and deformation of the extracellular matrix (ECM). Cells embedded within the ECM can provide mechanical resistance to this deformation through cell-matrix adhesion, helping to preserve the tissue's microstructure [17].
- Impact on Mechanical Properties: Freezing can alter the mechanical strength of tissues. For instance, one study found that freezing degenerated human annulus fibrosus tissue led to a significant decrease in peel stiffness and strength compared to fresh tissue, whereas non-degenerated bovine tissue showed no significant difference, suggesting that pre-existing tissue defects exacerbate freezing damage [23].
Diagram 1: Biophysical Pathways of Osmotic Dehydration During Slow Freezing. The chart outlines the cascade of events from initial ice formation to potential cellular outcomes, highlighting protective cryoprotectant (CPA) mechanisms.
Quantitative Data and Experimental Observations
The theoretical principles of osmotic dehydration are validated and quantified through empirical studies across various biological systems. The data below summarize key findings on mass transfer, cellular parameters, and functional outcomes.
Table 1: Mass Transfer Kinetics During Osmotic Dehydration of Frozen vs. Fresh Mango (Peleg's Model Parameters)
| Mango Sample Condition | Peleg's Rate Constant (k₁) for Water Loss (h·g/g i.w.c.) | Peleg's Capacity Constant (k₂) for Water Loss (g/g i.w.c.) | Equilibrium Water Content (Yₑ) (g/g i.w.c.) | Solid Gain at Equilibrium (g/g i.d.m.) |
|---|---|---|---|---|
| Fresh | 0.24 ± 0.02 | 0.24 ± 0.01 | 0.16 | 0.45 |
| Slow Frozen (-18°C) | 0.35 ± 0.03 | 0.29 ± 0.01 | 0.13 | 0.55 |
| Quick Frozen (-40°C) | 0.31 ± 0.03 | 0.28 ± 0.01 | 0.14 | 0.53 |
Source: Adapted from [21]. i.w.c. = initial water content; i.d.m. = initial dry matter. Note: Lower k₁ indicates faster initial water loss. Frozen samples showed slower initial water loss but higher final solid uptake compared to fresh mango, due to microstructural damage from ice crystals.
Table 2: Osmotic Properties of T Cells Relevant to Cryopreservation Protocol Design
| Cell Type | Permeable CPA | Temperature | Membrane Hydraulic Permeability (Lp) (μm/min/atm) | CPA Membrane Permeability (Ps) (cm/min) | Activation Energy (Eₐ) for Lp (kcal/mol) |
|---|---|---|---|---|---|
| Jurkat (T-cell line) | Me₂SO | 22°C | 0.40 | 2.2 × 10⁻⁴ | 10.2 |
| Jurkat (T-cell line) | Glycerol | 22°C | 0.43 | 5.3 × 10⁻⁵ | 12.8 |
| Primary Human T Cells | Me₂SO | 22°C | 0.51 | 2.3 × 10⁻⁴ | 9.6 |
| Primary Human T Cells | Glycerol | 22°C | 0.56 | 5.6 × 10⁻⁵ | 12.5 |
Source: Data compiled from [19]. These parameters are crucial for calculating the optimal cooling rate and CPA addition/removal times to minimize osmotic shock.
Table 3: Impact of Freezing on Tissue Mechanical Properties
| Tissue Type | Condition | Peel Stiffness | Peel Strength | Peel Toughness |
|---|---|---|---|---|
| Degenerated Human Annulus Fibrosus | Fresh | 100% (Baseline) | 100% (Baseline) | 100% (Baseline) |
| Degenerated Human Annulus Fibrosus | Frozen (-20°C for 3 weeks) | ↓ 50% | ↓ 37% | ↓ 41% (trend) |
| Non-Degenerated Bovine Annulus Fibrosus | Fresh | 100% (Baseline) | 100% (Baseline) | 100% (Baseline) |
| Non-Degenerated Bovine Annulus Fibrosus | Frozen (-20°C for 3 weeks) | No Significant Difference | No Significant Difference | No Significant Difference |
Source: Adapted from [23]. Freezing significantly compromises the interlamellar matrix properties of degenerated tissues, suggesting fresh testing is preferable for accurate mechanical assessment in such samples.
Experimental Protocols and Methodologies
Protocol: Investigating Freezing-Induced Deformation in Engineered Tissues
This protocol uses Cell Image Deformetry (CID) to quantify freezing-induced tissue deformation [17].
Tissue Preparation:
- Prepare a collagen solution (e.g., 3 mg/ml final concentration) in a chamber slide.
- Embed fluorescent markers (e.g., quantum dot-labeled MCF7 cells or fluorescent microspheres) at desired concentrations (e.g., 2×10⁵ particles/ml).
- Allow the engineered tissue (ET) to polymerize at 37°C for 1 hour. Add culture medium and incubate for 24 hours.
Freezing Setup:
- Place the ET on a temperature-controlled stage with two reservoirs creating a temperature gradient (e.g., -20°C to 4°C across a 6 mm gap).
Image Acquisition and Analysis:
- Capture successive fluorescence images at regular intervals (e.g., 1 second) during freezing.
- Use cross-correlation software (e.g., DaVis) to calculate deformation rates (u, v) in the x and y directions across interrogation windows.
- Compute the dilatation (e), a measure of tissue expansion or contraction, using the formula: e = ∂u/∂x + ∂v/∂y.
Protocol: Determining Osmotic Parameters via Flow Imaging Microscopy
This protocol details the use of Flow Imaging Microscopy (FIM) to determine the osmotic response of cells to CPA addition, a critical step for designing cryopreservation protocols [19].
Cell Preparation:
- Harvest and concentrate cells (e.g., Jurkat or primary T cells) to a standard density (e.g., 1×10⁶ cells/ml).
- Optionally, stain cells with Trypan Blue to differentiate viable from dead cells based on morphology.
Osmotic Challenge:
- Mix the cell suspension with hypertonic CPA solutions (e.g., containing Me₂SO or glycerol) in a FIM instrument's flow cell.
- The instrument automatically captures bright-field images of thousands of cells as they are exposed to the anisotonic medium.
Data Processing and Modeling:
- Software analyzes the images to calculate changes in cell volume over time.
- The volume-time data is fitted using a two-parameter formalism (2P) to solve the coupled differential equations for water and solute transport.
- The fitting yields the critical osmotic parameters: membrane hydraulic permeability (Lp) and CPA permeability (Ps).
Diagram 2: Experimental Workflow for Osmotic Parameter Determination. The flowchart outlines the key steps in using Flow Imaging Microscopy (FIM) to quantify cell membrane permeability, essential for designing optimized freezing protocols.
Protocol: Cryopreservation of Spheroids with Zwitterion/DMSO Solutions
This protocol describes slow-freezing using a novel CPA combination for complex multicellular systems [18].
CPA Solution Preparation:
- Prepare the optimized zwitterion/DMSO cryopreservation solution (e.g., ZD-10/15: 10 wt% zwitterion, 15 wt% DMSO, 75 wt% water). Ensure complete dissolution.
Spheroid Treatment:
- Transfer spheroids into the CPA solution and incubate for a sufficient equilibration time (determined empirically) at a non-toxic temperature (e.g., 4°C).
Slow-Freezing Process:
- Cool the samples in a controlled-rate freezer or a -80°C freezer using an insulated container (e.g., Mr. Frosty) to achieve an approximate cooling rate of -1°C/min.
- Store the frozen samples in liquid nitrogen vapor or at -80°C for long-term preservation.
Thawing and Assessment:
- Rapidly thaw samples in a 37°C water bath.
- Gently remove the CPA solution by dilution and centrifugation.
- Assess cell recovery and viability immediately post-thaw and after 24 hours of culture to avoid "false positive" results from temporarily stabilized cells.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagents and Materials for Investigating Osmotic Dehydration in Slow Freezing
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating Cryoprotectant | Standard CPA for cell lines and spheroids; often used in combination with other agents [18]. |
| Sucrose & Trehalose | Non-Permeating Cryoprotectants | Extracellular stabilizers that increase osmolarity and protect membrane integrity [19] [22]. |
| Synthetic Zwitterions (e.g., OE2imC3C) | Novel Non-Permeating CPA | Used with DMSO to synergistically protect spheroids and tissues via slow-freezing [18]. |
| Betaine | Zwitterionic Osmoprotectant & CPA | Incorporated into Lipid Nanoparticles (LNPs) during freezing to enhance stability and post-thaw mRNA delivery efficacy [22]. |
| Collagen Matrix (Type I) | Engineered Tissue Scaffold | Used to create 3D in vitro tissue models for studying freezing-induced deformation [17]. |
| Cal-520 AM Dye | Intracellular Calcium Indicator | Used in fluorescence microscopy to image calcium response states (OSCARS) under osmotic stress [24]. |
| Flow Imaging Microscope | Particle Analysis Instrument | High-throughput sizing and morphological analysis of cells during osmotic volume changes; allows viability discrimination [19]. |
| Controlled-Rate Freezer | Programmable Freezing Apparatus | Provides precise, reproducible control over cooling rates for optimizing slow-freezing protocols [20]. |
Osmotic dehydration is a central and inescapable phenomenon in slow freezing, governed by well-defined biophysical principles. The interplay between cooling rate, solute concentration, and cellular permeability dictates the survival and functionality of cells, tissues, and complex biologics. While the challenges of solute damage and mechanical stress are significant, advanced CPA strategies—including the use of synergistic permeating and non-permeating agents like zwitterions—offer powerful tools to mitigate these effects.
The future of optimizing slow-freezing protocols lies in the precise, quantitative understanding of system-specific osmotic parameters. The experimental methodologies outlined herein, particularly those leveraging high-throughput technologies like Flow Imaging Microscopy, provide a pathway to this precision. By integrating these fundamental principles with robust experimental data, researchers can rationally design cryopreservation protocols that transcend mere viability, preserving and even enhancing the critical functions of advanced therapeutic agents and biological constructs.
Mechanical Stresses on Plasma Membranes from Ice Crystals and Cellular Deformation
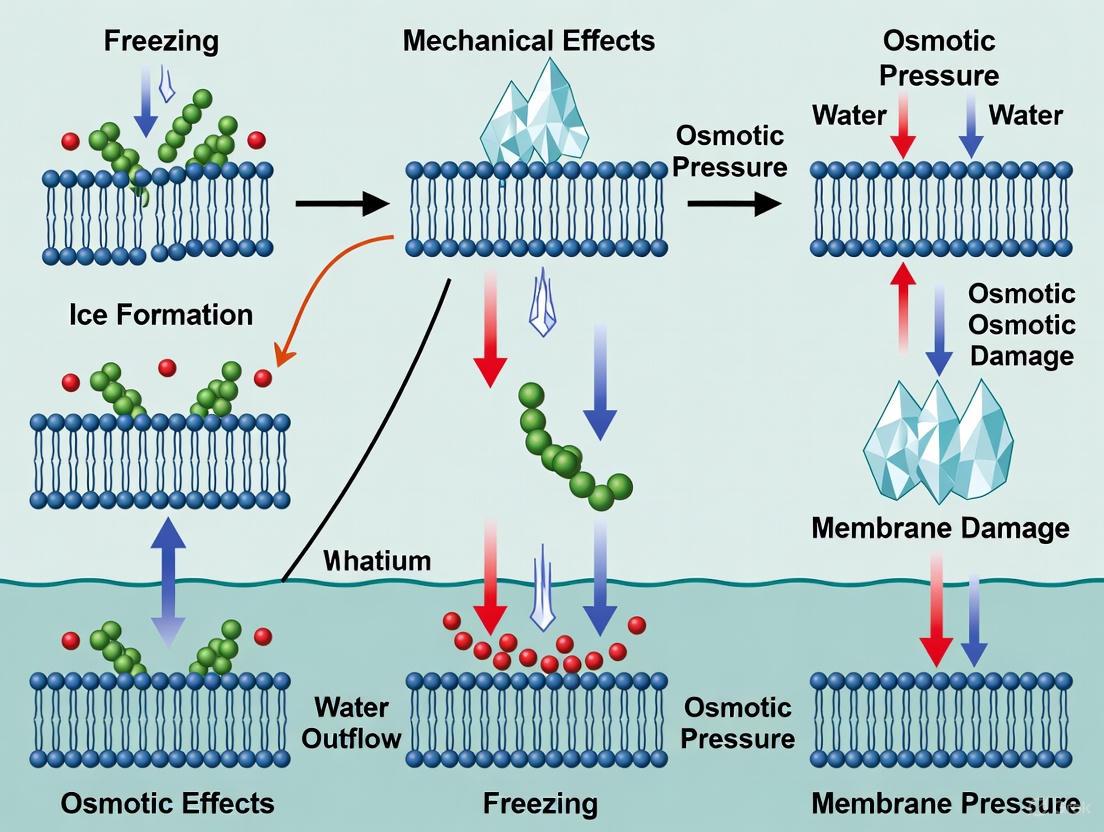
Freezing presents a complex biophysical challenge to cellular systems, combining mechanical and osmotic stresses that can compromise plasma membrane integrity and lead to cell death. This whitepaper synthesizes current research on the mechanisms of freeze-induced membrane stress, examining how ice crystal formation generates direct mechanical deformation while simultaneously triggering osmotically-driven water transport. We detail the specific cellular injury modes including expansion-induced lysis, loss of osmotic responsiveness, and freeze-induced vesicular structure formation. Experimental data from plant, mammalian, and engineered tissue models provide quantitative insights into membrane permeability, tension thresholds, and deformation patterns. The findings presented herein inform improved cryopreservation protocols, biopreservation strategies, and therapeutic approaches for cold-related tissue injuries, offering researchers in drug development and regenerative medicine a comprehensive technical framework for navigating freezing-induced cellular damage.
When biological systems encounter subzero temperatures, the resulting extracellular ice formation initiates a cascade of biophysical events that impose significant stress on cellular structures, particularly the plasma membrane. This stress manifests through two primary, interconnected pathways: direct mechanical deformation from growing ice crystals and indirect osmotic effects from freeze-concentrated solutes. The plasma membrane serves as the critical interface facing these challenges, with its integrity determining cellular survival post-thaw. Understanding these mechanisms is essential for numerous biomedical applications, from cryopreservation of cells for therapeutic use to managing cold-related injuries in tissues.
The mechanical and osmotic effects of freezing are intrinsically linked. Extracellular ice formation preferentially excludes solutes, creating a hypertonic extracellular environment that drives osmotic water efflux from cells [25]. This dehydration reduces cell volume, increasing intracellular solute concentration and potentially causing damaging phase transitions in membrane lipids. Simultaneously, the physical presence of ice crystals mechanically deforms both the extracellular matrix and cellular structures, creating compression and shear forces that can exceed the membrane's mechanical strength [17] [26]. The interplay between these mechanical and osmotic stresses creates a complex injury landscape that varies with cooling rate, temperature, and cell type.
Within the broader context of freezing effects on cells, this whitepaper specifically focuses on the mechanical consequences for plasma membranes. We examine how ice crystals directly deform cellular structures, how membranes respond to freeze-induced tension, and how cells can be engineered or treated to enhance membrane resilience during freezing. The insights provided aim to equip researchers with both fundamental knowledge and practical methodologies for investigating and mitigating freezing-induced membrane damage.
Mechanisms of Mechanical and Osmotic Stress
Extracellular Ice Crystals and Mechanical Deformation
The formation of extracellular ice crystals during freezing generates substantial mechanical stress on plasma membranes through multiple mechanisms. Ice crystals growing in extracellular spaces create physical barriers that compress cells, effectively reducing the available space and deforming cellular morphology [26]. This compression occurs as cells become confined within the narrowing spaces between advancing ice fronts, leading to membrane stretching, bending, and shear stresses.
The mechanical interaction between ice crystals and cells can be modeled as parallel plate compression. Computational analyses of this scenario reveal that compression generates both expansion tension (Te) and shear tension (Tsh) in the membrane, with expansion tension identified as the primary cause of cell lysis [26]. When cells are compressed between ice crystals, the membrane undergoes substantial expansion strain, particularly in regions adjacent to contact points with ice crystals. The resulting tension can reach levels sufficient to cause membrane failure, especially when combined with osmotic stresses.
The physical damage from ice crystals varies significantly with freezing rate. At slow cooling rates, large, needle-like ice crystals form extensively in extracellular spaces, creating widespread mechanical deformation across tissue architectures [27]. In contrast, rapid cooling promotes the formation of numerous small ice crystals both inside and outside cells, creating a different mechanical stress profile characterized by more uniform but potentially equally damaging forces.
Freeze-Induced Dehydration and Osmotic Stress
Concurrent with direct mechanical deformation, extracellular ice formation initiates profound osmotic stress on cells. As water freezes extracellularly, solutes become excluded from the growing ice lattice, creating a hypertonic unfrozen fraction that draws water out of cells through osmosis [25]. This freeze-induced dehydration reduces cell volume, concentrates intracellular solutes, and alters membrane morphology.
The osmotic response follows the Boyle-van't Hoff relationship for perfect osmometers, where cell volume is inversely proportional to external osmolality [28]. As cells lose water to the extracellular environment, their volume decreases, and the plasma membrane must accommodate this change in surface area. This can lead to membrane infolding or, in extreme cases, membrane collapse and irreversible damage. The rate of dehydration is critical – if too rapid, it can create steep osmotic gradients that overwhelm membrane water permeability; if too slow, it prolongs exposure to concentrated intracellular solutes.
The combination of mechanical and osmotic stress creates a particularly challenging environment for plasma membranes. Mechanical compression from ice crystals occurs simultaneously with osmotically-driven volume changes, creating complex stress patterns that challenge the membrane's structural integrity and can lead to various failure modes.
Cellular Injury Modes and Protective Responses
Membrane Failure Mechanisms
Plasma membranes respond to freezing stresses through several well-characterized failure mechanisms, each with distinct structural manifestations and functional consequences:
Expansion-Induced Lysis (EIL) occurs during thawing when previously dehydrated cells rapidly take up water and swell. If the membrane has been compromised or cannot accommodate the rapid expansion, the cells burst. In protoplasts from non-acclimated Arabidopsis leaves, EIL accounts for 19-28% of freezing injury at temperatures between -2°C and -4°C [25]. This form of injury is particularly associated with the formation of large endocytotic vesicles during freezing or osmotic dehydration that cannot be reincorporated into the plasma membrane during thawing.
Loss of Osmotic Responsiveness (LOR) manifests as the inability of cells to regulate volume changes in response to osmotic gradients after freezing and thawing. This injury stems from transitions in membrane lipid phases, specifically from lamellar (Lα) to hexagonal II (HII) phases, resulting from close apposition of the plasma membrane and internal endomembranes during dehydration [25]. This phase transition compromises membrane barrier function, leading to uncontrolled solute leakage and irreversible damage.
Freeze-Induced Vesicular Structures (FIVs) represent a protective response observed in cold-acclimated Arabidopsis protoplasts, where mechanical stress from ice crystal contact triggers immediate formation of vesicular structures that internalize portions of the plasma membrane [25]. These FIVs are subsequently reincorporated during thawing, effectively regulating membrane surface area and mitigating mechanical stress. FIV formation depends on extracellular calcium concentration, suggesting involvement of tension-activated calcium channels in this protective mechanism.
Surface Area Regulation and Membrane Adaptation
Cells possess remarkable ability to regulate plasma membrane surface area as a protective mechanism against freezing-induced mechanical stress. This surface area regulation (SAR) involves controlled endocytosis and exocytosis in response to membrane tension fluctuations [25]. When mechanical stress increases membrane tension, surface area is added through exocytosis of intracellular vesicles; conversely, decreased tension triggers endocytic retrieval of excess membrane.
In cold-acclimated plant cells, SAR manifests through specialized structures including exocytotic extrusions and freeze-induced vesicular structures (FIVs). Exocytotic extrusions appear as filiform projections on the surface of dehydrated protoplasts from cold-acclimated winter rye, providing reversible membrane reservoirs that can be reincorporated during thawing [25]. FIVs form specifically in response to mechanical deformation from ice crystals rather than osmotic dehydration alone, highlighting their role in mitigating mechanical stress [25].
The molecular machinery underlying SAR involves conventional endocytic and exocytic processes, though the specific mechanisms in freezing tolerance remain partially characterized. The dependence of FIV formation on extracellular calcium suggests mechanosensitive calcium channels may initiate the membrane trafficking response to mechanical stress [25]. Cold acclimation appears to enhance this capability through modifications to membrane composition and organization of the underlying cytoskeleton.
Quantitative Data and Experimental Findings
Membrane Tension and Cell Viability Parameters
Research across multiple cell types has yielded quantitative insights into the relationship between membrane tension, deformation, and cell survival during freezing. Computational modeling of compression experiments provides particularly valuable data on tension thresholds associated with membrane failure.
Table 1: Membrane Tension Parameters from Compression Experiments
| Cell Type | Initial Membrane Tension | Maximum Expansion Tension (Te) | Critical Strain for Viability Loss | Reference |
|---|---|---|---|---|
| Prostate adenocarcinoma (PC-3) | 2.7 mN/m | ~12 mN/m at ε=0.7 | 70% reduction at ε=0.7 | [26] |
| Endothelial cells | 2.7 mN/m | ~15 mN/m at ε=0.7 | Significant reduction at ε>0.7 | [26] |
| HeLa cells | 2.73-3.62 mN/m | Not reported | Not reported | [26] |
| C2C12 myoblasts | Not reported | Traction stress: 58-125 Pa (hypertonic) | Recoverable after osmotic shock | [29] |
The data reveal that membrane tension increases non-linearly with compressive strain, with dramatic escalation beyond approximately 70% strain (ε=0.7) [26]. This threshold correlates with significant reductions in cell viability, suggesting that expansion tension rather than shear tension serves as the primary determinant of membrane failure. The initial tension present in adhered cells (typically 2.7-3.6 mN/m) provides a baseline that influences how cells respond to additional stresses during freezing.
Cell Volume and Deformation Metrics
The osmotic component of freezing stress produces characteristic volume changes that vary with experimental conditions and cell type. These volumetric responses provide insight into membrane permeability and resilience.
Table 2: Cell Volume and Deformation Parameters Under Osmotic Stress
| Experimental System | Volume Change | Time Scale | Recovery Capability | Reference |
|---|---|---|---|---|
| C2C12 hypertonic shock (500 mOsm) | Continuous shrinkage for ~116 s | 160 s to minimum volume | ~90% volume recovery | [29] |
| C2C12 hypotonic shock (200 mOsm) | Immediate swelling, then shrinkage | 100-160 s swelling phase | Gradual recovery | [29] |
| MCF7 breast cancer cells | Dilatation dependent on cell concentration | Freezing process | Pattern variation with cell density | [17] |
| Plant protoplasts | Shrinkage due to freeze-induced dehydration | Immediate with ice formation | FIV-mediated protection | [25] |
The dynamic relationship between cell volume and membrane traction force reveals important mechanical behavior. Under hypertonic conditions, cell shrinkage correlates with increased traction stress (from 58 Pa to 125 Pa in C2C12 cells), while swelling decreases traction stress [29]. This inverse relationship demonstrates how osmotic volume changes directly influence mechanical interactions with substrates and presumably with ice crystals during freezing.
Experimental Methodologies
Cell Image Deformetry for Freezing-Induced Tissue Deformation
Cell Image Deformetry (CID) provides a powerful approach for quantifying freezing-induced deformation in engineered tissues and cellular constructs. This methodology enables spatial and temporal mapping of deformation patterns during controlled freezing protocols.
Protocol Overview:
- Sample Preparation: Engineered tissues are created by embedding quantum dot-labeled cells (e.g., MCF7 breast cancer cells) or fluorescent microspheres in a collagen matrix (typically 3 mg/ml final concentration). The constructs are allowed to polymerize at 37°C for 1 hour before adding culture medium and incubating for 24 hours [17].
Freezing Setup: Samples are placed on a temperature-controlled stage with two independently controlled temperature reservoirs separated by a 6 mm gap. Reservoirs are typically maintained at -20°C and 4°C to establish a controlled temperature gradient across the sample [17].
Image Acquisition: Successive fluorescence images are captured during freezing using a fluorescence macro/microscope with 2× magnification. Images are obtained at 1-second intervals using a high-sensitivity CCD camera [17].
Deformation Analysis: Acquired images are cross-correlated at 10-second intervals and divided into 32 × 32 pixel interrogation windows using specialized software (e.g., DaVis 7.1). The software calculates deformation rates in x and y directions at each window, which are then used to compute dilatation using the formula: e = ∂u/∂x + ∂v/∂y, where u and v represent deformation rates in the x and y directions, respectively [17].
This methodology enables researchers to quantify how different cell concentrations, matrix compositions, and freezing parameters influence tissue-scale deformation during ice formation.
Cryomicroscopy for Cellular Water Transport Measurement
Cryomicroscopy allows direct visualization and quantification of water transport across plasma membranes during freezing, providing critical parameters for modeling cellular response.
Protocol Overview:
- Cell Preparation: Cells in suspension are labeled with appropriate fluorescent markers if needed for visualization. Cell concentrations typically range from 2×10^5 to 8×10^5 cells/ml depending on experimental requirements [17].
Freezing Stage Configuration: Cells are placed on a specialized temperature-controlled stage (e.g., Linkam MD S600) capable of precise temperature regulation during freezing and thawing cycles [17].
Image Acquisition: A microscope (e.g., Olympus BX51) equipped with a CCD camera (e.g., Retiga 2000 R) captures images throughout the freezing process, documenting cell volume changes and intracellular ice formation if present [17].
Parameter Estimation: Membrane permeability parameters are estimated by comparing observed volume changes with mathematical models of water transport, typically using the approach of Mazur or similar formalism [17].
This methodology provides direct measurement of the fundamental biophysical parameters that govern cellular response to freezing, particularly the kinetics of water efflux during extracellular ice formation.
Parallel Plate Compression for Ice Crystal Simulation
Parallel plate compression experiments simulate the mechanical stress that cells experience between growing ice crystals, allowing controlled investigation of deformation-induced damage.
Protocol Overview:
- Cell Preparation: Target cells (e.g., human prostatic adenocarcinoma PC-3 cells) are suspended in solutions with varying osmotic concentrations to establish different initial cell sizes (e.g., 15.4, 17.8, and 20.5 μm diameters) [26].
Compression Test: Individual cells are compressed between parallel plates while measuring force and displacement. The compression rate is controlled to simulate different freezing conditions [26].
Viability Assessment: Cell viability post-compression is determined using standard assays (e.g., membrane integrity dyes) and correlated with compression parameters [26].
Computational Modeling: Experimental results are interpreted using computational models that treat the cell surface as an elastic membrane with specific Young's modulus and Poisson's ratio, and intracellular components as volume-maintaining elements [26].
This approach provides quantitative relationships between compressive strain, membrane tension, and cell survival, offering insights into the mechanical failure thresholds of plasma membranes under ice-like confinement.
The Scientist's Toolkit
Essential Research Reagents and Materials
Table 3: Key Reagents and Materials for Freezing-Stress Research
| Item | Function/Application | Example Use |
|---|---|---|
| Quantum Dot Labels (Qtracker 655) | Fluorescent cell labeling for tracking | Cell Image Deformetry of engineered tissues [17] |
| Type I Rat Tail Collagen | Engineered tissue scaffold | 3 mg/ml final concentration for CID samples [17] |
| FM1-43 Lipophilic Dye | Plasma membrane staining | Visualization of membrane behavior during freezing [25] |
| Phosphatidylcholine Diunsaturated Species | Membrane lipid modification | Enhancing freezing tolerance in protoplasts [25] |
| Extracellular Calcium Modulators | Manipulating calcium-dependent processes | Investigating FIV formation mechanisms [25] |
| Temperature-Controlled Stages (Linkam) | Precise thermal regulation | Cryomicroscopy and controlled freezing protocols [17] |
| Microspheres (20 μm diameter) | Osmotically-inactive cell analogs | Decoupling osmotic vs. mechanical effects [17] |
Signaling and Response Pathways
The cellular response to freezing-induced membrane stress involves complex signaling and mechanical pathways that integrate osmotic and mechanical stimuli into protective biological responses.
The diagram illustrates how freezing stresses initiate both mechanical and osmotic signaling pathways that converge on protective responses. Mechanical deformation from ice crystals and osmotic water efflux both contribute to increased membrane tension, which activates calcium influx through mechanosensitive channels [25]. This calcium signaling triggers cytoskeletal rearrangements and activates membrane trafficking machinery for surface area regulation through FIV formation and other mechanisms. Concurrently, changes in membrane composition during cold acclimation enhance resistance to phase transitions and maintain barrier function under dehydration stress [25]. The balance between these protective pathways and the intensity of the stress signals determines whether cells survive freezing or succumb to expansion-induced lysis or loss of osmotic responsiveness.
The investigation of mechanical stresses on plasma membranes from ice crystals and cellular deformation reveals a complex interplay between physical forces and biological responses. The plasma membrane serves as the primary interface where freezing injury is initiated, through both direct mechanical deformation from ice crystals and indirect osmotic effects from freeze-concentrated solutes. Understanding these mechanisms provides critical insights for developing improved cryopreservation protocols, protecting tissues from cold injury, and designing biostabilization strategies for pharmaceutical applications.
Key findings demonstrate that membrane lipid composition, surface area regulation mechanisms, and calcium-mediated signaling pathways collectively determine cellular resilience to freezing stress. The quantitative parameters presented herein – including membrane tension thresholds, deformation limits, and permeability characteristics – provide researchers with essential reference data for designing experiments and interpreting results. The experimental methodologies detailed offer standardized approaches for investigating freezing-induced membrane stress across different cell types and conditions.
As research in this field advances, emerging techniques in membrane biophysics, molecular biology, and computational modeling will further elucidate the intricate mechanisms of freeze-induced membrane damage and protection. This knowledge will ultimately enhance our ability to preserve cellular integrity under freezing conditions, with significant implications for regenerative medicine, pharmaceutical development, and fundamental cell biology.
This technical guide examines two critical freeze-induced lesions in cellular membranes: the lamellar-to-hexagonal II (Lα-HII) phase transition and expansion-induced lysis (EIL). Through detailed analysis of biophysical mechanisms and experimental findings, we explore how freeze-induced dehydration and mechanical stresses destabilize membrane integrity, with particular focus on the plasma membrane and chloroplast envelope. The content synthesizes current understanding of how cold acclimation, lipid composition alterations, and specific cryoprotective strategies mitigate these damaging transitions, providing researchers with comprehensive methodological frameworks for investigating membrane cryobehavior. Within the broader context of freezing effects on cells, this review establishes the fundamental role of membrane phase transitions in freezing injury and survival mechanisms, offering critical insights for cryopreservation protocol development and cold-tolerance engineering in biological systems.
When cells are exposed to subzero temperatures, extracellular ice formation initiates a cascade of biophysical events that ultimately lead to membrane destabilization. The plasma membrane serves as the primary barrier and sensor of freezing stress, making it particularly vulnerable to two distinct but interrelated forms of injury. The lamellar-to-hexagonal II phase transition represents a fundamental change in membrane lipid organization from a bilayer to a non-bilayer configuration, disrupting membrane integrity and functionality [30] [25]. Concurrently, expansion-induced lysis occurs as a consequence of osmotic excursions during freeze-thaw cycles, where irreversible reduction in plasma membrane surface area leads to membrane rupture during thawing [25]. These phenomena are not mutually exclusive; rather, they represent competing injury mechanisms that manifest across different temperature ranges and freezing conditions.
The investigation of these membrane lesions has profound implications for multiple fields, including cryopreservation, cryosurgery, and the development of freeze-tolerant organisms. In cryopreservation, unintended membrane phase transitions significantly reduce post-thaw viability of cells and tissues [31] [3]. Conversely, in cryosurgical applications, precisely inducing these transitions contributes to targeted destruction of tumor cells [31]. Understanding the molecular mechanisms governing these processes therefore enables both the prevention and targeted induction of membrane failure, depending on the application.
Fundamental Mechanisms of Membrane Phase Transitions
Lamellar-to-Hexagonal II (Lα-HII) Phase Transition
The lamellar (Lα) phase represents the natural bilayer organization of membrane lipids, characterized by a planar structure with polar head groups oriented toward the aqueous interface and hydrophobic tails forming the membrane interior. Under specific conditions, including dehydration, certain lipids can undergo a transition to the hexagonal II (HII) phase, wherein lipid molecules form cylindrical inverted micelles arranged in a hexagonal pattern [32]. This reorganization creates aqueous channels surrounded by lipid head groups, with hydrocarbon chains extending outward [33].
This phase transition is energetically favorable for specific lipids with intrinsic negative curvature, particularly phosphatidylethanolamine (PE) and monogalactosyldiacylglycerol (MGDG) [32] [34]. The molecular shape of these lipids, characterized by relatively small head groups compared to their hydrocarbon chains, promotes the formation of curved structures essential for HII phase formation. The transition proceeds through proposed stalk intermediates that facilitate the connection between opposing membrane bilayers [32]. These transient structures represent the initial step in membrane fusion and phase transition pathways, with their stability determining the kinetics of the Lα-HII transition.
Freeze-induced dehydration provides the primary trigger for this transition in biological systems. As extracellular ice forms, water is progressively removed from membrane surfaces, increasing the concentration of solutes and effectively dehydrating the membrane interface [30]. This dehydration brings opposing membranes into close apposition (often plasma membrane and chloroplast envelope in plant cells), creating the conditions necessary for the Lα-HII transition [30] [34]. The resulting HII phases disrupt membrane integrity, leading to loss of osmotic responsiveness and solute leakage [25].
Expansion-Induced Lysis (EIL)
Expansion-induced lysis represents a mechanical failure of the plasma membrane resulting from irreversible reduction in surface area during freeze-thaw cycles. During freezing, osmotic contraction of the cell causes endocytotic vesiculation of the plasma membrane, internalizing portions of the membrane as vesicles [25] [34]. If the reduction in surface area exceeds a critical threshold, the membrane cannot accommodate the volumetric expansion during thawing, resulting in lysis [34].
The incidence of EIL is highly dependent on the cooling rate and the lipid composition of the membrane. At slow cooling rates, extensive dehydration occurs, promoting greater endocytotic vesiculation and consequently higher susceptibility to EIL [25]. Membranes with higher proportions of diunsaturated phospholipids, such as phosphatidylcholine species, demonstrate reduced EIL incidence due to improved flexibility and capacity for surface area regulation [25].
Table 1: Comparative Characteristics of Membrane Lesions in Freezing Injury
| Parameter | Lamellar-to-Hexagonal II Transition | Expansion-Induced Lysis |
|---|---|---|
| Primary cause | Freeze-induced dehydration and close membrane apposition | Osmotic contraction and endocytotic vesiculation |
| Temperature range | Below -4°C (in plant protoplasts) | -2°C to -4°C (in plant protoplasts) |
| Membrane outcome | Loss of bilayer continuity, phase change | Irreversible reduction in surface area |
| Cellular manifestation | Loss of osmotic responsiveness | Membrane rupture during thawing |
| Key influencing factors | Lipid composition, membrane proximity | Cooling rate, membrane elasticity |
Experimental Evidence and Quantitative Data
Temperature Dependence and Survival Signatures
Studies using protoplast models have revealed distinct "survival signatures" that reflect the temperature dependence of different injury mechanisms. In protoplasts isolated from non-acclimated Arabidopsis thaliana leaves, survival decreases sharply at specific temperature thresholds corresponding to the dominance of different injury mechanisms [34]. Research demonstrates that EIL predominates in the range of -2°C to -4°C, while LOR-HII becomes the primary injury mechanism below -4°C [34].
Quantitative analysis of protoplast survival reveals the specific contribution of each lesion. In one study, constitutive expression of the COR15a gene in Arabidopsis thaliana resulted in decreased survival (6-12% lower than wild-type) in the -2°C to -4°C range, but increased survival (16% for wild-type vs. 44% for transgenic at -5.5°C) at lower temperatures [34]. This paradoxical effect was explained by the gene's dual impact: increasing susceptibility to EIL while providing protection against Lα-HII transitions.
Table 2: Quantitative Effects of Cold Acclimation and Genetic Modification on Freezing Tolerance
| Experimental Condition | EIL Incidence | Lα-HII Transition Incidence | Overall Survival |
|---|---|---|---|
| Non-acclimated protoplasts | High (19-28% injury at -2°C to -4°C) | High at <-4°C | Low |
| Cold-acclimated protoplasts | Reduced | Significantly reduced | High |
| COR15a expression (non-acclimated) | Increased | Significantly reduced | Variable (temperature-dependent) |
| Increased diunsaturated phosphatidylcholine | Reduced | Reduced | High |
| Artificial reduction of nucleation temperature | Not applicable | Increased | Reduced |
Membrane Composition and Phase Behavior
Fourier transform infrared spectroscopy (FTIR) studies have provided molecular-level insights into freezing effects on membrane phase behavior. Research on LNCaP prostate tumor cells demonstrates that the ice nucleation temperature significantly affects membrane lipid organization [31]. When ice nucleates at higher temperatures (-3°C), membranes undergo a highly cooperative liquid crystalline to gel phase transition with low residual conformational disorder. In contrast, nucleation at lower temperatures (-10°C) results in less cooperative transitions with higher conformational disorder [31].
The extent of cellular dehydration directly correlates with membrane structural changes. Cryomicroscopy and FTIR studies show that reduced lipid hydration under dehydrating conditions correlates strongly with cellular volumetric decreases [35]. This dehydration-induced membrane stress is considered a key component of "solution effects" injury in cryobiology, distinct from intracellular ice formation [35].
Methodologies for Investigating Membrane Phase Transitions
Freeze-Fracture Electron Microscopy (FFEM)
Freeze-fracture electron microscopy has been instrumental in identifying and characterizing membrane phase transitions in frozen specimens. The standard protocol involves:
- Sample Preparation: Isolate protoplasts or cells and suspend in appropriate medium. For plant studies, protoplasts are typically isolated from leaves by enzymatic digestion [30] [34].
- Freezing Protocol: Cool samples at controlled rates (typically 1°C/min) to target subzero temperatures. Hold at specified temperatures for fixed durations (e.g., 90 minutes for protoplast studies) [34].
- Cryofixation: Rapidly freeze samples using liquid nitrogen-cooled propane or ethane to preserve membrane structures.
- Fracture and Replication: Fracture samples under vacuum at -150°C and shadow with platinum/carbon to create replicas.
- Electron Microscopy: Examine replicas using transmission electron microscopy to visualize membrane architecture.
This technique has revealed various intermediate structures in the Lα-HII transition, including aparticulate lamellae and inverted micellar intermediates [30] [32].
Fourier Transform Infrared Spectroscopy (FTIR)
FTIR provides a powerful method for monitoring real-time changes in membrane phase behavior during freezing. The standard experimental approach includes:
- Sample Preparation: Concentrate cells (e.g., LNCaP prostate tumor cells) into pellets between CaF2 infrared windows separated by 6μm spacers [31].
- Temperature Control: Mount samples in a variable temperature cell regulated by liquid nitrogen cooling and electronic temperature controller.
- Ice Nucleation Control: Use ice nucleators like Pseudomonas syringae or controlled nucleation with cooled copper wire to achieve specific nucleation temperatures (-3°C to -10°C) [31].
- Spectral Acquisition: Collect infrared spectra during cooling and warming cycles (typically 2°C/min) over temperature ranges from -80°C to +90°C.
- Data Analysis:
- Monitor CH2 symmetric stretching band (~2850 cm⁻¹) for membrane phase behavior
- Analyze amide-I, -II, and -III bands for protein secondary structure
- Track water/ice phases using absorption bands between 2680-1950 cm⁻¹
This methodology has revealed that membrane phase transitions coincide with ice nucleation temperature and cellular dehydration state [31].
³¹P-Nuclear Magnetic Resonance (NMR) Spectroscopy
³¹P-NMR provides direct detection of lipid phase transitions through characteristic chemical shift patterns:
- Sample Preparation: Prepare lipid dispersions (10% w/v) in appropriate buffers. For studies on chloroplast envelope lipids, use mixtures of MGDG, DGDG, SQDG, PC, and PG at native ratios [34].
- Temperature Equilibration: Place samples in NMR spectrometer at initial temperature (e.g., 0°C) and equilibrate.
- Spectral Acquisition: Collect ¹H-decoupled ³¹P-NMR spectra at temperature increments (1-5°C), allowing 10 minutes equilibration at each temperature.
- Phase Identification:
- Lamellar phases produce asymmetric lineshapes with high-field shoulder
- Hexagonal II phases produce reversed asymmetry with low-field shoulder
- Isotropic phases produce narrow symmetric resonances
This technique has demonstrated that COR15am polypeptide increases the Lα-HII phase transition temperature of DOPE and promotes lamellar phase formation in chloroplast envelope lipid mixtures [34].
Diagram 1: Pathways of Freezing-Induced Membrane Injury
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents for Investigating Membrane Phase Transitions
| Reagent/Material | Function/Application | Example Use |
|---|---|---|
| Protoplast isolation enzymes | Cell wall digestion for membrane access | Isolation of plant protoplasts for freeze-fracture studies [30] |
| Dioleoylphosphatidylethanolamine (DOPE) | Model lipid for HII phase formation | In vitro studies of Lα-HII transition kinetics [34] |
| Dimethyl sulfoxide (DMSO) | Cryoprotectant for membrane stabilization | Preventing phase transitions in cryopreservation [3] |
| Pseudomonas syringae | Ice nucleator for controlled freezing | Standardizing ice nucleation temperature in FTIR studies [31] |
| FM1-43 fluorescent dye | Membrane staining for cryomicroscopy | Visualizing plasma membrane behavior during freezing [25] |
| COR15am polypeptide | Cryoprotective protein | Investigating modulation of Lα-HII transition temperature [34] |
| Trehalose | Non-permeating cryoprotectant | Membrane stabilization during dehydration [3] |
Cold Acclimation and Protective Mechanisms
Cold acclimation induces multiple physiological changes that protect membranes against freeze-induced phase transitions. Key adaptive mechanisms include:
Lipid Composition Modifications
Cold-acclimated plants exhibit significant alterations in plasma membrane lipid composition, particularly increased proportions of diunsaturated species of phosphatidylcholine [25]. This modification decreases the propensity for both Lα-HII transitions and EIL by altering the intrinsic curvature of membrane lipids and increasing membrane flexibility [25]. Similar changes in lipid composition have been observed in cold-acclimated winter rye, spring oat, and Arabidopsis thaliana [25].
Surface Area Regulation and Freeze-Induced Vesicular Structures
Cold-acclimated plant cells develop specialized mechanisms for surface area regulation (SAR) that mitigate mechanical stresses during freezing. In protoplasts from cold-acclimated Arabidopsis leaves, freezing induces the formation of freeze-induced vesicular structures (FIVs) that appear immediately after ice crystals contact the protoplast surface [25]. These structures are incorporated back into the plasma membrane during thawing, providing a mechanism to accommodate volume changes without permanent surface area loss.
The formation of FIVs is dependent on extracellular calcium concentration and is thought to involve conventional endo- and exocytotic processes [25]. This mechanism represents a biological response to mechanical stress that complements the physicochemical adaptations in membrane composition.
Cryoprotective Proteins and Peptides
Specific polypeptides induced during cold acclimation directly modulate membrane phase behavior. The COR15am polypeptide, targeted to chloroplasts in Arabidopsis thaliana, increases the Lα-HII phase transition temperature of dioleoylphosphatidylethanolamine and promotes lamellar phase formation in chloroplast envelope lipid mixtures [34]. This action defer freeze-induced HII phase formation to lower temperatures (lower hydrations), thereby protecting membrane integrity during freezing.
Diagram 2: Protective Mechanisms Against Membrane Freezing Injury
The study of lamellar-to-hexagonal II phase transitions and expansion-induced lysis provides fundamental insights into the primary mechanisms of freezing injury in biological systems. These phenomena represent the interface between thermodynamic constraints and biological adaptation, where membrane composition and organization determine survival outcomes under freezing conditions.
Understanding these processes has enabled significant advances in cryopreservation protocols, particularly in optimizing cooling rates and cryoprotectant formulations to avoid both destructive phase transitions and mechanical membrane failure [3]. Furthermore, this knowledge provides strategic approaches for engineering freeze-tolerant organisms through manipulation of membrane lipid composition and expression of cryoprotective polypeptides.
Future research directions include elucidating the specific molecular mechanisms of calcium-dependent FIV formation, developing more precise methods for quantifying phase transition intermediates, and engineering synthetic membrane systems with enhanced resistance to freeze-induced damage. The continued investigation of membrane phase transitions will undoubtedly yield new strategies for managing cellular responses to freezing stress across medical, agricultural, and biotechnological applications.
The Role of Surface Area Regulation in Freezing Tolerance
Freezing tolerance is a critical survival trait for cells and organisms exposed to subzero temperatures. While biochemical adaptations have been extensively studied, the mechanical and osmotic stresses generated by ice formation present equally fundamental challenges. This technical guide examines the underappreciated yet crucial role of surface area regulation (SAR) as a cellular defense mechanism against freezing injury. Within the broader context of mechanical and osmotic effects research, SAR represents a fundamental process whereby cells dynamically manage their plasma membrane area to withstand freeze-induced deformations.
During extracellular freezing, cells face a dual assault: osmotic dehydration as liquid water is sequestered as ice, and mechanical stress from growing ice crystals [25]. The plasma membrane, as the primary interface, must accommodate these drastic physical changes without rupturing. Surface area regulation enables cells to maintain membrane integrity through controlled addition or removal of membrane material, serving as a critical adaptation that complements osmotic and cryoprotectant-based strategies in freezing tolerance.
Theoretical Framework: Surface Area Regulation Mechanisms
Fundamental Principles of Surface Area Regulation
Surface area regulation constitutes a discrete cellular task distinct from volume or shape regulation [36]. Eukaryotic cells maintain substantial endomembrane reservoirs that can be rapidly mobilized to accommodate surface area fluctuations. The membrane tension hypothesis posits that cells detect and respond to deviations from a membrane tension set point, triggering membrane trafficking events to restore homeostatic tension levels [36].
Two primary models explain how cells regulate surface area during mechanical stress:
- Membrane Unfolding Model: Pre-existing membrane reservoirs in the form of microvilli, wrinkles, or invaginations unfold to provide additional surface area
- Exocytosis Model: Intracellular vesicles fuse with the plasma membrane to deliver new lipid bilayer
These mechanisms are not mutually exclusive; cells may employ both strategies depending on the magnitude and duration of surface area requirements [37].
Membrane Trafficking in Freezing Stress
During freezing, SAR occurs through specialized membrane trafficking events. In cold-acclimated Arabidopsis protoplasts, freeze-induced vesicular structures (FIVs) form immediately upon ice crystal contact [25]. These FIVs are distinct from endocytotic vesicles induced by osmotic dehydration and appear specifically responsive to mechanical deformation. During thawing, FIVs reincorporate into the plasma membrane, demonstrating the reversible nature of this SAR mechanism [25].
The molecular machinery governing these processes likely involves conventional endocytic and exocytic pathways, with calcium signaling playing a modulatory role. Experimental evidence indicates FIV formation depends on extracellular calcium concentration, suggesting potential involvement of calcium channels gated by membrane tension [25].
Quantitative Data on Surface Area Changes During Freezing
Table 1: Documented Surface Area Changes in Biological Systems Under Stress
| Cell Type/System | Stress Condition | Surface Area Change | Regulatory Mechanism | Citation |
|---|---|---|---|---|
| Cold-acclimated Arabidopsis protoplasts | Extracellular freezing | Formation of freeze-induced vesicular structures (FIVs) | Membrane internalization | [25] |
| Dictyostelium cells | Cell division | ~20% increase during cytokinesis | Exocytosis of intracellular vesicles | [38] |
| Mammalian bladder epithelium | Cyclical expansion | >100% area increase | Unfolding + membrane insertion | [37] |
| Plant protoplasts | Freeze-induced dehydration | Endocytotic vesicle formation | Membrane internalization | [25] |
Table 2: Key Membrane Properties Relevant to Freezing Tolerance
| Parameter | Value/Range | Biological Significance | Experimental System | |
|---|---|---|---|---|
| Membrane physical stretch capacity | 2-3% maximum | Explains necessity for membrane trafficking | Lipid bilayer studies | [38] |
| Apparent surface area occupied by membrane reservoirs | 21-130% | Buffer for rapid surface expansion | Various eukaryotic cells | [38] |
| FIV formation dependence on extracellular calcium | Concentration-dependent | Suggests mechanosensitive channel involvement | Arabidopsis protoplasts | [25] |
| Lipid composition change during cold acclimation | Increased diunsaturated phosphatidylcholine | Resistance to Lα-to-HII phase transition | Winter rye, spring oat, Arabidopsis | [25] |
Experimental Approaches and Methodologies
Model Systems for SAR Studies
Research into surface area regulation during freezing employs diverse model systems:
- Plant protoplasts: Isolated from cold-acclimated Arabidopsis leaves, enabling direct membrane visualization [25]
- Dictyostelium cells: Flattened by agar overlay to eliminate membrane reservoir effects [38]
- Winter cereals: Including barley and rye, for physiological studies of freezing tolerance [39]
Key Methodological Approaches
Membrane Visualization and Area Quantification
The agar overlay method enables precise surface area measurement by physically suppressing membrane reservoirs. Cells are flattened under an agar sheet, eliminating microvilli and wrinkles that complicate surface area calculations [38]. Following flattening, surface area can be quantified through:
- Optical sectioning fluorescence microscopy with z-intervals of 0.2-0.3μm
- Deconvolution processing to remove out-of-focus light
- Cell outline analysis from dorsoventral and lateral areas
Alternatively, fluorescent lipid analogs (e.g., FM1-43) permit direct membrane labeling and tracking. When combined with confocal fluorescence cryomicroscopy, this approach enables real-time visualization of membrane dynamics during freezing [25].
Freezing Tolerance Assessment
Standardized protocols for evaluating freezing tolerance include:
- Controlled freezing regimes: Typically decreasing at 2°C/h from 0°C to target temperatures [39]
- Regrowth assessment (FT-R): Visual estimation of recovery after freezing [39]
- Electrolyte leakage measurements: Quantifying membrane integrity by ion leakage [39]
- Chlorophyll fluorescence parameters: Assessing photosystem integrity in plants [39]
Research Reagent Solutions
Table 3: Essential Research Reagents for SAR and Freezing Tolerance Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| Membrane Labels | FM1-43, fluorescent lipid analogs | Visualizing membrane dynamics, endocytosis, and recycling | Live-cell imaging during freezing [25] [38] |
| Cryoprotectants | Dimethyl sulfoxide (DMSO), trehalose | Membrane stabilization, reducing cryo-damage | Cryopreservation protocols [13] [40] |
| Ion Channel Modulators | Calcium concentration manipulations | Investigating calcium-dependent FIV formation | Mechanosensing studies [25] |
| Cytoskeletal Drugs | Thiabendazole (microtubule depolymerizer) | Probing cytoskeletal contributions to SAR | Cell division and shape studies [38] |
| Lipid Composition Tools | Diunsaturated phosphatidylcholine species | Artificially modifying membrane fluidity | Freezing tolerance enhancement [25] |
Signaling and Regulatory Pathways
The following diagram illustrates the integrated signaling network through which cells perceive freezing stress and activate surface area regulation mechanisms:
Experimental Workflow for SAR Studies
The diagram below outlines a comprehensive methodology for investigating surface area regulation in freezing tolerance:
Surface area regulation represents a fundamental mechanism in freezing tolerance that complements established osmotic and cryoprotectant strategies. Through controlled membrane trafficking—including freeze-induced vesiculation, exocytosis, and reservoir unfolding—cells dynamically manage their surface area to withstand mechanical and osmotic stresses during freezing.
The implications for drug development and cryopreservation are substantial. Understanding SAR mechanisms could inform:
- Improved cryopreservation protocols for biological specimens, tissues, and organs [13]
- Novel therapeutic approaches for enhancing cellular survival in cryomedicine
- Biomimetic strategies for stabilizing synthetic membrane systems in biotechnology
Future research should focus on elucidating the molecular machinery of membrane tension sensing and the signaling pathways coordinating SAR with other freezing tolerance mechanisms. As climate change increases the frequency of freeze-thaw events [39], understanding these fundamental cellular processes becomes increasingly critical for both agricultural and medical applications.
Advanced Preservation Technologies: From Slow Freezing to Vitrification in Biomedical Applications
Principles and Procedures of Slow Freezing Cryopreservation
Slow freezing cryopreservation represents a fundamental methodology in biotechnology and regenerative medicine for the long-term storage of biologically active constructs. This technical guide delineates the core principles underpinning this technique, with specific focus on its role in mitigating the mechanical and osmotic stresses that cells encounter during freezing. The procedure leverages controlled cooling rates and cryoprotective agents (CPAs) to promote cellular dehydration, thereby minimizing lethal intracellular ice formation (IIF) [41] [42]. We provide a comprehensive examination of the thermodynamic and mechanical effects on cells, summarized quantitative data, detailed experimental protocols, and essential research tools. When executed with precision, slow freezing provides a robust mechanism for preserving a wide array of cell types and tissues, facilitating advanced research and clinical applications in drug development.
Cryopreservation is the process of preserving organelles, cells, and tissues at very low temperatures, typically below -130°C, where all biological activity effectively ceases [41] [43]. The fundamental challenge lies in navigating the phase change of water from liquid to solid without inducing catastrophic damage to the cellular architecture. Slow freezing is a primary cryopreservation strategy characterized by a controlled, gradual reduction in temperature, typically at rates between -0.5°C/min to -3°C/min [44] [45]. This method is theoretically and practically useful for counteracting the cell's natural response to ice formation, which includes osmotic shock, membrane damage, and ice crystal formation that can lead to cell death [41].
The core mechanism of slow freezing is controlled cellular dehydration [44] [45]. As the extracellular solution freezes, the formed ice is relatively pure, excluding solutes and thereby increasing the solute concentration in the remaining unfrozen liquid. This creates an osmotic gradient that draws water out of the cell across the semi-permeable plasma membrane. A sufficiently slow cooling rate provides the time necessary for this water efflux to occur, thus minimizing supercooling and preventing the nucleation of ice inside the cell, which is almost always lethal [41] [42]. The success of this process is therefore a balance between the cooling rate and the cell's intrinsic permeability to water, ensuring that cellular volume contraction keeps pace with the freezing front [3].
Mechanical and Osmotic Effects of Freezing on Cells
Understanding the cellular damage mechanisms during freezing is paramount to developing effective preservation protocols. The injuries can be broadly categorized into two interconnected theories: osmotic stress and mechanical damage from ice crystals.
Osmotic Stress and "Solution Effects"
As extracellular ice forms, solutes are excluded from the crystal lattice, leading to a dramatic increase in the concentration of electrolytes and other solutes in the unfrozen fraction. Cells exposed to these high solute concentrations face "solution effects," which can cause protein denaturation, lipid membrane restructuring, and irreversible damage to critical enzymes due to altered ionic interactions [42]. Furthermore, the osmotic imbalance causes rapid efflux of water, leading to excessive cell shrinkage that can exceed the minimum tolerated cell volume, causing membrane lysis [3] [42].
Mechanical Effects of Ice Crystals
The physical presence of ice crystals presents a direct mechanical threat. Extracellular ice can mechanically disrupt the fine structures of tissues and cellular junctions [42]. However, the most critical damage arises from intracellular ice formation (IIF). IIF is widely used as an indicator of cell death, as the ice crystals can rupture organelles and the plasma membrane itself [41] [42]. The probability of IIF is highly dependent on the cooling rate; slow cooling minimizes this risk by promoting dehydration, whereas overly rapid cooling does not allow sufficient time for water to leave the cell, resulting in supercooling and eventual intracellular nucleation [3].
The following diagram illustrates the critical pathways and outcomes for cells undergoing a slow freezing process, highlighting the key decision points that determine survival or death.
Critical Parameters and Optimization Data
The efficacy of slow freezing is governed by several interdependent parameters that must be optimized for specific cell types. The table below summarizes key quantitative data and their impacts on cryopreservation outcomes.
Table 1: Key Parameters for Optimizing Slow Freezing Cryopreservation
| Parameter | Typical Range / Examples | Impact & Rationale | Key References |
|---|---|---|---|
| Cooling Rate | -0.5°C/min to -3°C/min (general);~1°C/min (HSCs, MSCs);Rapid cooling (oocytes, pancreatic islets) | Slow rate allows cellular dehydration, preventing IIF; optimal rate is cell-type specific. | [3] [46] [44] |
| Permeating CPAs | DMSO (1.5 M);Glycerol (1.5 M);Ethylene Glycol | Penetrate cell, depress freezing point, reduce "solution effects" by diluting electrolytes. Low toxicity is crucial. | [41] [3] [46] |
| Non-Permeating CPAs | Sucrose (0.1-0.15 M);Trehalose;Hydroxyethyl Starch | Increase extracellular osmolarity, promoting gentle dehydration. Synergistic with permeating CPAs. | [3] [46] [45] |
| Cell Viability Post-Thaw | ~70-80% (MSCs, general);Varies by cell type and protocol | Benchmark for protocol success; requires post-thaw functional assays. | [44] [45] |
| Seeding Step | Manual nucleation at -8°C | Triggers controlled extracellular ice formation, preventing destructive supercooling. | [46] |
The choice and concentration of CPAs are particularly critical. The table below provides a comparative analysis of commonly used agents.
Table 2: Comparison of Common Cryoprotective Agents (CPAs)
| Cryoprotective Agent | Type | Mechanism of Action | Relative Toxicity | Common Applications |
|---|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Permeating | Depresses freezing point, penetrates membrane, reduces electrolyte concentration. | Moderate | Cultured mammalian cells, hematopoietic stem cells, testicular tissue. |
| Glycerol | Permeating | Similar to DMSO; first discovered CPA. | Lower (but lower efficacy) | Microorganisms, spermatozoa. |
| Ethylene Glycol (EG) | Permeating | Lower molecular weight, faster penetration. | Moderate (similar to DMSO) | Often used in vitrification mixtures. |
| Sucrose | Non-Permeating | Increases extracellular osmolarity, promoting dehydration. | Low | Used as an additive with permeating CPAs (e.g., DMSO). |
| Trehalose | Non-Permeating | Stabilizes membranes in dry state; naturally occurring in freeze-tolerant organisms. | Low | Difficult to introduce into mammalian cells; used extracellularly. |
Detailed Experimental Protocol for Controlled Slow Freezing
The following protocol for the controlled slow freezing of testicular tissues [46] and mesenchymal stem cells (MSCs) [44] [45] exemplifies a robust, clinically relevant methodology. This procedure can be adapted for other cell types with appropriate optimization of CPA composition and cooling rates.
Reagent Preparation
- Freezing Medium: Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) or Hank's Buffered Salt Solution (HBSS) supplemented with 1.5 M DMSO, 0.1 M to 0.15 M sucrose, and 10 mg/mL Human Serum Albumin (HSA) [46]. The medium should be prepared sterilely and chilled on ice before use.
- Equipment: Programmable freezer (e.g., Planer Kryo 360), isopropyl alcohol freezing container (e.g., "Mr. Frosty") for uncontrolled rate freezing, cryogenic vials, liquid nitrogen storage system, personal protective equipment (PPE) for handling liquid nitrogen.
Pre-Freezing: Sample Preparation and CPA Equilibration
- Harvest and Suspend: Harvest the target cells (e.g., via trypsinization for adherent cells) or dissect tissues into small fragments (e.g., ~4x4x4 mm³). Create a single-cell suspension or prepare tissue fragments in a suitable basal medium [46].
- CPA Addition and Equilibration: Gently mix the cell pellet or tissue fragments with the pre-chilled freezing medium. The equilibration period is critical for CPA penetration and osmotic balance. Incubate the mixture on ice for 30 minutes [46]. Gently agitate periodically to ensure uniform exposure.
The Controlled Freezing Process
- Transfer to Cryovials: Aliquot the cell/tissue suspension into labeled cryogenic vials.
- Programmable Freezing Cycle: Place the vials in the programmable freezer and initiate the following controlled cooling cycle [46]:
- Start at 0°C.
- Cool at -1°C/min to -8°C. This initial slow cooling begins the dehydration process.
- Hold at -8°C for 10 minutes. Perform manual seeding during this hold by briefly touching the vials with forceps pre-cooled in liquid nitrogen. This step triggers controlled extracellular ice crystallization, preventing destructive supercooling.
- Resume cooling at -0.5°C/min to -40°C. This very slow rate allows for extensive dehydration.
- Cool rapidly at -7°C/min to -70°C.
- Long-Term Storage: Immediately transfer the cryovials from the programmable freezer to a liquid nitrogen storage tank, either in the vapor phase (around -150°C) or the liquid phase (-196°C) for long-term preservation [46] [44].
Thawing and Post-Thaw Processing
- Rapid Thawing: Retrieve the vial from liquid nitrogen and immediately place it in a 37°C water bath with gentle agitation. Thawing is complete when only a small ice crystal remains (approximately 2 minutes) [46] [44]. Using a water bath carries a contamination risk, so consider using alternative dry heating equipment for greater safety [44].
- CPA Removal and Washing: Immediately after thawing, transfer the cell suspension to a tube containing pre-warmed (37°C) culture medium supplemented with HSA or serum. This dilution step is crucial to reduce CPA toxicity. Gently mix and centrifuge to pellet the cells. Aspirate the supernatant containing the toxic CPAs.
- Resuspension and Viability Assessment: Resuspend the cell pellet in fresh, complete culture medium. Plate the cells and assess viability using a dye exclusion method (e.g., Trypan Blue). It is essential to perform functional assays post-thaw, as immediate viability can be a "false positive," with apoptotic activity leading to cell loss after 24 hours of culture [18] [44].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Slow Freezing Cryopreservation
| Item / Reagent | Function / Application | Example & Notes |
|---|---|---|
| Programmable Freezer | Provides precise, reproducible control over cooling rates. Critical for protocol development and GMP compliance. | Planer Kryo 360 series. Alternative: inexpensive isopropyl alcohol containers (e.g., Nalgene "Mr. Frosty") for less critical applications. |
| Permeating CPA (DMSO) | Primary penetrating cryoprotectant. Reduces ice crystal formation and mitigates solute effects. | USP grade DMSO (e.g., CryoSure) is recommended for clinical-grade protocols [46]. |
| Non-Permeating CPA (Sucrose) | Osmotic balancer; enhances dehydration and reduces the required concentration of toxic permeating CPAs. | Often used in combination with DMSO in a typical 1.5 M DMSO / 0.1 M sucrose formulation [46]. |
| Serum Albumin | Supplements freezing medium; helps stabilize cell membranes and mitigate CPA toxicity. | Human Serum Albumin (HSA) is used in clinical-grade protocols to avoid xenogenic components [46]. |
| Commercial Freezing Media | Ready-to-use, standardized formulations that can simplify the process and ensure consistency. | CELLBANKER series (e.g., serum-free, xeno-free CHEMICAL defined versions for stem cells) [41]. CultureSure (Fujifilm) [18]. |
| Novel CPA (Zwitterions) | Emerging class of cell-impermeable CPAs that strongly interact with water, inhibiting ice crystal growth. | Imidazolium/carboxylate zwitterions used in research, showing synergistic effects with DMSO for spheroid and tissue cryopreservation [18]. |
Slow freezing cryopreservation remains a cornerstone technique for the preservation of cells and tissues in biomedical research and clinical therapy. Its success hinges on a deep understanding of the osmotic and mechanical stresses imposed on cells during freezing—primarily lethal intracellular ice formation and damaging solute concentration effects. By meticulously optimizing parameters such as cooling rate and CPA composition, researchers can effectively steer cells toward a pathway of dehydration and survival. The detailed protocols and tools outlined in this guide provide a foundation for robust and reproducible cryopreservation, a process that is indispensable for the advancement of cell-based assays, biobanking, and the burgeoning field of regenerative medicine.
Vitrification represents a radical advancement in cryopreservation technology, enabling the transition of aqueous solutions directly into a glassy amorphous solid without the destructive formation of ice crystals. This process stands in stark contrast to conventional slow-freezing methods, which rely on controlled ice crystal formation and growth. Within the context of research on mechanical and osmotic effects of freezing on cells, vitrification addresses two fundamental sources of cryoinjury: the mechanical shearing of cellular structures by ice crystals and the osmotic stress induced by solute concentration during phase separation. By achieving a glassy state through extremely high cooling rates and high concentrations of cryoprotective agents (CPAs), vitrification bypasses ice crystallization entirely, thereby preserving cellular integrity and function to an unprecedented degree.
The principle of vitrification hinges on rapidly increasing solution viscosity during cooling until molecular motion effectively ceases, forming a metastable glass that maintains the molecular disorder of a liquid. For researchers and drug development professionals, understanding and optimizing vitrification protocols is critical for preserving sensitive biological systems—from individual cells to complex tissues—that would otherwise be damaged by conventional freezing methods. This technical guide explores the fundamental mechanisms, protocols, and applications of vitrification, with particular emphasis on its role in mitigating mechanical and osmotic cellular damage.
Fundamental Principles of Vitrification
Thermodynamic Basis of Glass Formation
Vitrification achieves a non-equilibrium state through ultra-rapid cooling that prevents the nucleation and growth of ice crystals. When an aqueous solution undergoes cooling, the temperature eventually reaches the point where ice would normally form. However, if the cooling rate is sufficiently high and the CPA concentration adequate, the viscosity increases dramatically until the system solidifies into an amorphous glass. The critical cooling rate required to achieve vitrification depends on several factors, including the composition of the solution, the concentration of cryoprotectants, and the volume of the sample.
The glass transition temperature (Tg') represents a critical thermodynamic parameter where the supercooled liquid transforms into a glassy solid. Differential scanning calorimetry studies of freezing medium containing Leibovitz L-15 medium with 4 mg/mL human serum albumin (HSA), 1.5M DMSO, and 0.1M sucrose revealed a glass transition temperature of -120.49°C, with crystallization occurring at -20°C when cooled at 2.5°C/min and melting at -4.11°C [47]. Understanding these thermal properties is essential for designing effective vitrification and warming protocols that maintain the glassy state throughout the cryopreservation process.
Comparative Analysis: Vitrification vs. Slow Freezing
Table 1: Comparison of Vitrification and Slow Freezing Methodologies
| Parameter | Vitrification | Slow Freezing |
|---|---|---|
| Cooling Rate | Extremely rapid (minutes) | Very slow (hours) |
| CPA Concentration | High (30-50%) | Low (1-2M) |
| Ice Formation | Eliminated | Controlled formation |
| Equipment Needs | Simple (liquid nitrogen) | Programmable freezer |
| Theoretical Basis | Non-equilibrium freezing | Equilibrium freezing |
| Primary Damage Mechanisms | CPA toxicity, osmotic stress | Ice crystal formation, solute effects |
| Survival Rates (Oocytes) | >90% [48] | ~66% [48] |
| Clinical Pregnancy Rates (Embryos) | 40.5% [49] | 21.4% [49] |
The superiority of vitrification is demonstrated across multiple biological systems. In human cleavage stage embryos, vitrification achieved a survival rate of 96.9% compared to 82.8% with slow freezing, with significantly higher rates of embryos maintaining excellent post-warming morphology (91.8% vs. 56.2%) [49]. Similarly, clinical pregnancy rates (40.5% vs. 21.4%) and implantation rates (16.6% vs. 6.8%) were substantially higher in the vitrification group [49].
Mechanisms of Cell Damage and Protection
Mechanical and Osmotic Effects in Conventional Freezing
During conventional slow freezing, ice formation initiates in the extracellular space, concentrating solutes in the remaining unfrozen solution. This creates an osmotic gradient that draws water out of cells, leading to cellular dehydration and volumetric changes. If cooling occurs too slowly, excessive dehydration causes "solution effects" injury from concentrated solutes. If cooling occurs too rapidly, intracellular ice forms, causing mechanical damage to membranes and organelles.
The mechanical damage from ice crystals manifests as shearing of cellular membranes and disruption of subcellular structures, including microtubules and organelles [50]. Simultaneously, osmotic stress occurs as water shifts across membranes, creating pressure gradients that can exceed membrane integrity limits. These coupled mechanical-osmotic effects represent the primary sources of cryoinjury in conventional freezing protocols.
How Vitrification Mitigates These Effects
Vitrification addresses both mechanical and osmotic damage sources through fundamentally different physical mechanisms. By eliminating ice formation entirely, vitrification prevents mechanical shearing of cellular structures. The uniform glassy state maintains spatial relationships at the molecular level, preserving membrane integrity and organelle architecture.
The osmotic stress in vitrification is managed differently than in slow freezing. While vitrification requires higher CPA concentrations, which increases osmotic stress during addition and removal, this stress occurs at higher temperatures where cells are more metabolically active and better able to accommodate osmotic challenges. Additionally, the development of one-step warming protocols has demonstrated that simplified methods can effectively manage osmotic stress while maintaining high survival rates [50].
Molecular Stress Responses
Cells subjected to cryopreservation stressors activate protective molecular pathways. Heat shock protein 70 (HSP70) plays a particularly important role in cellular protection during vitrification. Temperature stress during vitrification procedures induces HSP70 expression, which functions as a molecular chaperone to assist protein folding under stress conditions [51].
Table 2: Cellular Stress Responses in Cryopreservation
| Stress Type | Cellular Response | Protective Mechanism |
|---|---|---|
| Temperature Stress | HSP70 induction [51] | Protein refolding, membrane stabilization |
| Oxidative Stress | Antioxidant enzyme activation | Reduction of reactive oxygen species |
| Osmotic Stress | Osmolyte synthesis | Intracellular-extracellular equilibrium |
| Apoptotic Signals | Caspase inhibition [51] | Prevention of programmed cell death |
HSP70 also plays a role in suppressing apoptosis in vitrified oocytes by inhibiting the activation of APAF-1, caspase 9, and caspase 3, thereby maintaining cell viability after warming [51]. This dual function of HSP70—protein chaperoning and apoptosis suppression—represents a crucial cellular adaptation to the combined temperature and osmotic stresses encountered during vitrification.
Experimental Protocols and Methodologies
One-Step vs. Multi-Step Warming Protocols
Recent advances in vitrification methodology have focused on simplifying warming procedures. A comprehensive study compared traditional multi-step warming with a simplified one-step protocol for vitrified-warmed blastocyst stage embryos [50].
One-Step Warming Protocol:
- Warming in 1M sucrose solution for 1 minute
- Total procedure time reduction >90% compared to multi-step
- Elimination of sequential sucrose concentration steps
Traditional Multi-Step Warming Protocol:
- Initial exposure to 1M sucrose for 1 minute
- Transfer to 0.5M sucrose for 3 minutes
- Final washing in solution for 10 minutes
This study demonstrated comparable survival rates between the two methods, with no significant differences in clinical pregnancy rates (42.6% vs. 44.3%) or ongoing pregnancy rates (33.2% vs. 37.5%) across 1402 transferred embryos from 989 patients [50]. The success of the simplified protocol suggests that careful optimization of osmotic conditions can maintain efficacy while significantly improving laboratory efficiency.
Optimized Freezing Protocol for Ovarian Tissue
For more complex tissues, optimized freezing protocols must account for tissue-specific thermodynamic properties:
Freezing Curve Parameters:
- Initial hold: 5 minutes at 4°C
- Cooling rate 1: 1°C/min to -7°C
- Seeding: 60°C/min to -32°C
- Cooling rate 2: 10°C/min to -15°C
- Cooling rate 3: 0.3°C/min to -40°C
- Final cooling: 10°C/min to -140°C
Thawing Protocol:
- Slow warming: 3.5-minute step in cold chamber to reach Tg'
- Rapid warming: 2-minute incubation at 37°C to reach Tm [47]
This protocol, developed through precise characterization of the freezing medium's thermal properties, demonstrates how understanding thermodynamic parameters enables optimization of vitrification procedures for specific biological materials.
Vitrification Workflow for Embryos
The following diagram illustrates the complete experimental workflow for embryo vitrification, comparing traditional and simplified approaches:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Vitrification Studies
| Reagent/Material | Function | Example Applications |
|---|---|---|
| Permeating CPAs (EG, DMSO, PG) | Penetrate cell membranes, depress freezing point | Embryo/oocyte vitrification [50] [52] |
| Non-Permeating CPAs (Sucrose, Trehalose) | Osmotic buffers, extracellular glass formers | All vitrification protocols [50] [6] |
| Macromolecular Additives (HSA, Ficoll) | Increase solution viscosity, reduce CPA toxicity | Ovarian tissue cryopreservation [47] |
| Specific Protein Supplements (HSP70) | Mitigate temperature stress, inhibit apoptosis | Experimental oocyte vitrification [51] |
| Base Media (Leibovitz L-15, MRS broth) | Maintain physiological pH and osmolarity | Ovarian tissue, probiotic vitrification [47] [6] |
| Vitrification Carriers (Cryotop, Cryoloop) | Enable ultra-rapid cooling | Embryo/oocyte vitrification [51] |
Quantitative Analysis of Vitrification Outcomes
Survival and Pregnancy Outcomes by Embryo Quality
Table 4: Pregnancy Outcomes by Embryo Quality and Warming Technique
| Embryo Quality | Warming Technique | Clinical Pregnancy Rate | Ongoing Pregnancy Rate |
|---|---|---|---|
| Top Quality (G1) | Multi-Step | 52.3% | 46.0% |
| Top Quality (G1) | One-Step | 54.6% | 48.1% |
| Good Quality (G2) | Multi-Step | 38.6% | 27.8% |
| Good Quality (G2) | One-Step | 40.0% | 33.0% |
Analysis of pregnancy outcomes by embryo morphology reveals that while top-quality embryos (G1) consistently outperform good-quality embryos (G2), both categories show comparable results between warming techniques [50]. This demonstrates that the simplified one-step protocol does not compromise outcomes across different embryo quality grades.
Impact of Developmental Stage
Embryos vitrified at different developmental stages show distinct survival potentials:
Day 5 Embryos:
- Multi-step: 44.8% clinical pregnancy rate, 35.3% ongoing pregnancy rate
- One-step: 46.5% clinical pregnancy rate, 40.0% ongoing pregnancy rate
Day 6 Embryos:
- Multi-step: 28.0% clinical pregnancy rate, 18.3% ongoing pregnancy rate
- One-step: 31.2% clinical pregnancy rate, 23.4% ongoing pregnancy rate [50]
These results confirm that developmental competence influences outcomes more significantly than the specific warming technique employed.
Apoptotic Signaling Pathways in Vitrified Cells
The cellular response to vitrification stresses involves complex signaling pathways that determine survival versus apoptotic outcomes:
This pathway illustrates how vitrification stressors can trigger apoptotic signaling through both mitochondrial and receptor-mediated pathways, while also highlighting the protective role of HSP70 in suppressing caspase activation and promoting cell survival [51].
Vitrification represents a paradigm shift in cryopreservation methodology, effectively addressing the fundamental mechanical and osmotic challenges that have limited the success of conventional freezing approaches. By achieving a glassy state without ice formation, vitrification preserves cellular integrity and function across diverse biological systems, from individual gametes to complex tissues.
The continued refinement of vitrification protocols—including simplified one-step warming methods and optimized CPA combinations—demonstrates the dynamic evolution of this technology. For researchers and drug development professionals, understanding the principles and practices of vitrification is essential for advancing cryopreservation applications while minimizing cellular damage. As our knowledge of cellular stress responses improves and protocol efficiency increases, vitrification will continue to expand the boundaries of what can be successfully preserved for research and clinical applications.
Cryopreservation is an indispensable technique in biomedical research and drug development, enabling the long-term storage of cells, tissues, and other biological materials. This process is fundamental to applications ranging from cell-based therapeutics to the maintenance of research cell lines. However, the freezing and thawing procedures inherent to cryopreservation induce significant stresses that can compromise cellular integrity and function. The core challenge lies in managing the mechanical and osmotic effects of ice formation, which pose a dual threat to cell survival [53] [3].
During freezing, the formation of intracellular and extracellular ice crystals can mechanically disrupt cellular membranes and organelles—a phenomenon known as mechanical damage [53] [54]. Concurrently, as pure water freezes, solutes in the remaining liquid phase become increasingly concentrated, creating a hypertonic environment that imposes severe osmotic stress on cells, leading to detrimental shrinkage and potential lysis [3] [54]. The foundational thesis of modern cryobiology is that successful preservation must address both of these injury pathways.
Cryoprotectant Agents (CPAs) are chemical compounds specifically employed to mitigate these damaging effects. Their primary role is to protect cell physiology, with a particular emphasis on maintaining the structure and function of the cellular membrane, a primary target of freezing injury [53]. While effective, most CPAs, particularly the penetrating varieties, introduce their own challenge: inherent cellular toxicity [53] [55]. Therefore, the use of CPAs represents a delicate balance between protection and toxicity. This guide provides an in-depth examination of the mechanisms by which CPAs stabilize cellular membranes and the advanced strategies being developed to mitigate their toxic effects, framed within the critical context of managing the mechanical and osmotic consequences of freezing.
Mechanisms of Freezing-Induced Cell Damage
To understand the protective role of cryoprotectants, one must first appreciate the two primary, interconnected mechanisms of freezing damage: osmotic stress and mechanical injury from ice crystals.
Osmotic Stress and "Solution Effects"
As the temperature drops below 0°C, extracellular ice begins to form. Because ice crystals exclude solutes, the concentration of dissolved ions (e.g., sodium, chloride) and other solutes in the unfrozen fraction of the solution increases dramatically. This creates a steep osmotic gradient across the cell membrane, causing water to efflux from the cell in an attempt to achieve equilibrium. This leads to cellular dehydration and excessive shrinkage, which can damage the plasma membrane and internal structures [3] [54]. These deleterious consequences of rising solute concentrations are collectively termed "solution effects" [54]. The extent of damage is directly correlated with the degree of solute concentration, and CPAs like glycerol are known to protect cells by colligatively reducing this concentration at any given sub-zero temperature [54].
Mechanical Damage from Ice Crystals
Intracellular ice formation (IIF) is almost universally lethal to cells [54]. When cooling is too rapid, water within the cell does not have sufficient time to exit and equilibrate with the external environment. Instead, it supercools and eventually freezes, forming ice crystals that can pierce and rupture membranes, organelles, and the cytoskeleton [53] [56]. The physical expansion of water upon freezing exacerbates this damage. Even when ice forms only extracellularly, large crystals can mechanically crush cells in densely packed suspensions or tissues [54]. Furthermore, during the thawing process, a phenomenon known as ice recrystallization can occur, where smaller ice crystals melt and re-freeze onto larger ones, increasing the average ice crystal size and causing additional mechanical stress [53].
The following diagram illustrates the sequential damage pathways triggered by freezing, from the initial physical event to the ultimate functional consequences for the cell.
Classification and Mechanisms of Action of Cryoprotectants
Cryoprotectants are broadly categorized based on their ability to cross the cell membrane, which dictates their primary mechanism of action. The following table summarizes the core characteristics of these two classes.
Table 1: Key Characteristics of Penetrating and Non-Penetrating Cryoprotectants
| Aspect | Penetrating (PA) | Non-Penetrating (NPA) |
|---|---|---|
| Molecular Size | Small (< 100 Da) [57] | Large (> 1,000 Da) [57] |
| Membrane Permeability | High [3] | None or very low [53] [57] |
| Location of Action | Intracellular and extracellular [53] | Extracellular only [53] [57] |
| Primary Mechanism | Colligatively depress freezing point, replace intracellular water, reduce ice formation [3] [58] | Increase extracellular osmolality, promote protective dehydration, inhibit ice recrystallization [53] [3] |
| Toxicity | Higher, especially at high concentrations and temperatures [53] [57] | Generally lower [53] [57] |
| Common Examples | DMSO, Glycerol, Ethylene Glycol, Propylene Glycol [53] [3] | Sucrose, Trehalose, Polyvinyl Alcohol, Hydroxyethyl Starch (HES) [53] [3] [58] |
Membrane Stabilization by Penetrating Cryoprotectants
Penetrating CPAs, such as Dimethyl Sulfoxide (DMSO) and glycerol, are small, neutral molecules that readily diffuse across the plasma membrane. Their protective action is multifaceted:
- Reduction of Intracellular Ice Formation: By entering the cell, PAs colligatively lower the freezing point of the intracellular solution. This increases the amount of "unfrozen" water at a given sub-zero temperature, dramatically reducing the probability of lethal intracellular ice formation during rapid cooling [3] [58].
- Buffering Osmotic Shock: During slow cooling, PAs mitigate the extent of cell shrinkage by partially replacing the water that exits the cell. This helps maintain cell volume above a critical minimum threshold, preventing irreversible membrane damage due to excessive contraction [58].
- Direct Membrane Interactions: Some PAs, like DMSO, are known to interact directly with membrane lipids. At low concentrations, DMSO can alter membrane dynamics and increase permeability, facilitating water efflux. It is also hypothesized to stabilize membrane proteins and structures by substituting for water molecules in hydrogen bonding, thereby preserving native conformations during dehydration [3].
Membrane Stabilization by Non-Penetrating Cryoprotectants
Non-penetrating CPAs are typically large molecules or polymers that remain outside the cell. They protect through extracellular mechanisms:
- Inducing Protective Dehydration: NPAs increase the extracellular osmolality, creating a controlled osmotic gradient that draws water out of the cell before freezing occurs. This purposeful dehydration reduces the amount of freezable water inside the cell, thus minimizing the potential for intracellular ice formation during subsequent rapid cooling [53] [3].
- Inhibiting Ice Recrystallization: Many NPAs, particularly polymers like Polyvinyl Alcohol (PVA) and hydroxyethyl starch (HES), function as potent ice recrystallization inhibitors (IRIs). They adsorb to the surface of ice crystals, preventing smaller crystals from merging into larger, more damaging ones during the thawing process [53] [59]. This is a critical mechanism for reducing mechanical damage to the cell membrane.
- Vitrification Enhancement: NPAs contribute to the overall solute concentration and increase the viscosity of the solution. This aids in achieving vitrification—the transition of the solution into a glassy, amorphous solid without ice crystallization—when used in combination with high concentrations of PAs [53] [3].
The synergistic interaction of both classes of cryoprotectants in stabilizing the cell membrane against mechanical and osmotic stress is illustrated below.
Toxicity of Cryoprotectants and Mitigation Strategies
A significant challenge in cryopreservation is the inherent toxicity of CPAs, particularly penetrating agents. DMSO, the most widely used CPA, exemplifies this dual nature of protection and toxicity [55].
Mechanisms of Toxicity
CPA toxicity manifests through several mechanisms:
- Membrane and Protein Damage: At high concentrations or with prolonged exposure, PAs can disrupt lipid bilayers, leading to membrane lysis [3]. They can also denature cellular proteins and interfere with enzyme function [53].
- Oxidative Stress: Exposure to CPAs can induce the generation of reactive oxygen species (ROS), leading to oxidative damage of lipids, proteins, and DNA [53].
- Altered Cellular Function: DMSO is known to influence cellular processes such as differentiation and can cause epigenetic changes, which is a significant concern for cell-based therapies [55]. Clinical side effects in patients receiving DMSO-cryopreserved stem cell infusions include nausea, cardiovascular effects, and, rarely, severe reactions like cardiac arrest [59].
- Osmotic Shock During Addition/Removal: The introduction and removal of CPAs, especially PAs, must be carefully controlled. Rapid changes in CPA concentration can cause severe osmotic swelling or shrinkage, leading to cell lysis or activation of apoptotic pathways [3].
Table 2: Common Cryoprotectants and Their Associated Toxicities
| Cryoprotectant | Class | Reported Toxicities | Notes on Usage |
|---|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating [53] | Alters differentiation, induces oxidative stress, clinical side effects (nausea, arrhythmia) [59] [55]. Can cause epigenetic changes [55]. | Gold standard but controversial. Use minimal effective concentration (e.g., 5-10%) [3]. |
| Glycerol | Penetrating [3] | Generally less toxic than DMSO but permeates cells more slowly [58]. | Commonly used for red blood cell cryopreservation [3]. |
| Ethylene Glycol | Penetrating [3] | Toxic metabolite profile; requires careful handling [58]. | Often used in vitrification mixtures for oocytes/embryos [3]. |
| Propylene Glycol | Penetrating [53] | Similar toxicity profile to other PAs [53]. | Used in clinical cryopreservation protocols [53]. |
| Sucrose / Trehalose | Non-penetrating [53] [3] | Very low toxicity. High concentrations can cause excessive hypertonicity [3]. | Often used in combination with PAs to reduce their required concentration [3]. |
| Polyvinyl Alcohol (PVA) | Non-penetrating [53] | Very low cytotoxicity [53]. | Effective ice recrystallization inhibitor [53]. |
Strategies for Toxicity Mitigation
Several strategies have been developed to minimize CPA toxicity while maintaining high post-thaw viability:
- Optimization of CPA Cocktails: Using combinations of permeating and non-permeating agents allows for a reduction in the concentration of any single, more toxic PA. For example, adding sucrose, trehalose, or polymers like HES can enable a decrease in DMSO concentration while maintaining or even improving cryoprotection [3] [58]. This approach leverages the synergistic effects of different CPAs [3].
- Stepwise Addition and Removal: CPA toxicity is concentration- and temperature-dependent [53]. Adding and removing CPAs in a stepwise manner at low temperatures (e.g., 0-4°C) slows down their chemical activity and gives cells time to equilibrate osmotically, thereby reducing both chemical and osmotic stress [3].
- Use of Natural and Low-Toxicity Agents: There is growing interest in identifying and applying natural cryoprotectants with lower toxicity profiles. These include antifreeze proteins (AFPs), sugars like trehalose, and natural deep eutectic systems (NADES) [53]. Recent research has even explored the cryoprotective efficacy of plant-based protein hydrolysates, such as those derived from grape seeds, which show promise in stabilizing biological membranes and reducing oxidation [60].
- Incorporation of Antioxidants: Adding antioxidants like melatonin to cryopreservation media can counteract the oxidative stress induced by CPAs and the freezing process itself. Melatonin has emerged as a potent cytoprotectant for gametes and somatic cells due to its antioxidant and gene-modulation properties [53].
- Development of DMSO-Free Formulations: Driven by regulatory and clinical concerns, significant effort is being invested in creating fully defined, serum-free, and DMSO-free cryopreservation solutions. These often rely on optimized mixtures of other PAs (e.g., propylene glycol) and NPAs [61] [59].
Experimental Protocols for Evaluation
To systematically evaluate the efficacy and toxicity of novel CPA formulations, researchers employ a suite of standardized assays. The following workflow outlines a typical experimental process for CPA screening and validation.
Protocol: Cryoprotectant Screening Assay
Objective: To compare the post-thaw viability and membrane integrity of cells cryopreserved in different CPA formulations.
Materials:
- Cell culture (e.g., adherent cell line like HEK293 or suspension cells like Jurkat).
- CPA stocks: High-purity DMSO, Ethylene Glycol, Sucrose, Trehalose, HES, etc. [57].
- Base medium: Serum-free or serum-containing culture medium.
- Equipment: Programmable controlled-rate freezer, water bath (37°C), hemocytometer or automated cell counter, flow cytometer with Annexin V/PI staining capability, centrifuge.
Method:
- CPA Formulation: Prepare the test CPA cocktails in base medium. Examples include:
- Control A: 10% DMSO in medium.
- Control B: Serum-free commercial cryopreservation medium.
- Test Formulation 1: 5% DMSO + 150mM Trehalose.
- Test Formulation 2: 10% Ethylene Glycol + 5% HES.
- Test Formulation 3: 8% Propylene Glycol + 200mM Sucrose. Filter sterilize all solutions (0.22 µm).
Cell Preparation and CPA Addition:
- Harvest cells in mid-log phase of growth. Determine viability and concentration.
- Pellet cells and resuspend in pre-chilled (4°C) CPA formulations at a defined concentration (e.g., 1x10^7 cells/mL).
- Aliquot cell suspensions into cryovials (e.g., 1 mL/vial).
Freezing and Storage:
- Place cryovials in a controlled-rate freezer. Use a standard slow-freezing protocol: e.g., cool from 4°C to -40°C at a rate of -1°C/min, then transfer rapidly to liquid nitrogen vapor phase for long-term storage [3].
- For vitrification studies, use high CPA concentrations and ultra-rapid cooling by direct plunging into liquid nitrogen [59].
Thawing and CPA Removal:
- Rapidly thaw cryovials in a 37°C water bath with gentle agitation.
- Immediately and gently transfer the cell suspension to a tube containing 10mL of pre-warmed culture medium to dilute the CPA.
- Centrifuge gently to pellet cells and remove the CPA-containing supernatant.
- Resuspend the cell pellet in fresh culture medium.
Post-Thaw Analysis (Conduct within 1-4 hours of thaw):
- Viability: Mix cell suspension with Trypan Blue dye (1:1) and count live (unstained) and dead (blue) cells on a hemocytometer. Calculate percentage viability.
- Membrane Integrity/Apoptosis: Use a flow cytometry-based assay with Annexin V and Propidium Iodide (PI) to distinguish live (Annexin V-/PI-), early apoptotic (Annexin V+/PI-), and late apoptotic/necrotic (Annexin V+/PI+) populations.
- Functional Assays: Depending on the cell type, perform specific functional tests 24-48 hours post-thaw (e.g., metabolic activity with MTT/XTT assay, clonogenic assays to measure proliferative capacity, or differentiation potential for stem cells).
The Scientist's Toolkit: Key Reagents and Materials
Table 3: Essential Research Reagents for Cryoprotectant Studies
| Reagent / Material | Function / Application | Notes |
|---|---|---|
| Dimethyl Sulfoxide (DMSO) | Penetrating cryoprotectant; the current gold standard for many cell types [3]. | Use high-purity, cell culture-tested grade. Final concentration typically 5-15% [3] [57]. |
| Trehalose | Non-penetrating disaccharide; stabilizes membranes via water replacement; antioxidant [3] [60]. | Often used at 50-250 mM in combination with PAs to reduce their toxicity [3]. |
| Polyvinyl Alcohol (PVA) | Synthetic polymer; potent ice recrystallization inhibitor (IRI) [53] [59]. | Used at low concentrations (0.5-2% w/v). Low cytotoxicity makes it attractive for DMSO-free formulations [53]. |
| Hydroxyethyl Starch (HES) | Non-penetrating polymer; contributes to extracellular vitrification and osmotic support [58]. | Common component in clinical cryopreservation solutions for hematopoietic stem cells [58]. |
| Annexin V / Propidium Iodide (PI) | Flow cytometry dyes for quantifying apoptosis and necrosis post-thaw [3]. | Critical for assessing membrane integrity and early signs of cryo-damage beyond simple viability. |
| Programmable Controlled-Rate Freezer | Equipment that provides a reproducible, linear cooling rate (e.g., -1°C/min) [3]. | Essential for standardizing slow-freezing protocols and comparing results across experiments. |
The successful cryopreservation of cells is a delicate balancing act between mitigating the mechanical and osmotic damage caused by ice formation and minimizing the inherent toxicity of the protective agents themselves. Penetrating cryoprotectants like DMSO protect primarily by entering the cell, depressing the freezing point, and buffering osmotic shifts, while non-penetrating agents act extracellularly to promote controlled dehydration and inhibit destructive ice recrystallization. The toxicity of traditional PAs remains a significant hurdle, particularly for clinical applications.
The future of cryoprotectant development lies in the rational design of multi-component, synergistic cocktails that leverage the strengths of different CPA classes while minimizing their individual drawbacks. This includes the incorporation of natural products, antioxidants, and advanced polymeric materials with specific ice-shaping properties. As research continues to refine our understanding of CPA interactions with the cellular membrane and the physical chemistry of freezing solutions, the goal remains the development of highly effective, low-toxicity, and potentially universal preservation protocols that ensure the functional integrity of biological materials for research and therapeutic use.
The successful cryopreservation of biological specimens represents a cornerstone of modern biotechnology, regenerative medicine, and pharmaceutical development. At the heart of this process lies a critical balancing act: optimizing the cooling rate to minimize two competing mechanisms of cellular injury. On one hand, excessively slow cooling exposes cells to prolonged hypertonic stress as extracellular ice formation concentrates solutes. On the other hand, overly rapid cooling prevents sufficient cellular dehydration, resulting in lethal intracellular ice formation [62]. This technical guide examines the fundamental principles and contemporary methodologies for cooling rate optimization, framed within the broader context of research on the mechanical and osmotic effects of freezing on cells.
The classical "two-factor hypothesis" of cryoinjury describes this inverse relationship between damage mechanisms, suggesting an optimal cooling rate that minimizes both solute effects and intracellular ice formation for each cell type [62]. Understanding this balance is not merely academic; it has direct implications for clinical applications, including the preservation of advanced therapy medicinal products (ATMPs) where post-thaw viability and functionality are critical to therapeutic efficacy [63]. Recent research has further elucidated how cooling rates influence the morphological features of freeze-concentrated solutions (FCS), where cells accumulate during freezing, providing new insights into the physical environment cells experience during cryopreservation [64].
Theoretical Foundations of Cryoinjury
Competing Mechanisms of Freezing Damage
When cells are subjected to subzero temperatures, they face two primary, interconnected pathways of injury:
Solute Effects (Slow Cooling Damage): During slow cooling, ice forms extracellularly first, excluding solutes from the crystal structure and concentrating them in the remaining liquid phase. This creates a hypertonic environment that draws water out of cells through osmosis, leading to excessive cell dehydration, membrane damage, and toxic solute concentrations [62]. The extent of dehydration is time-dependent, making this phenomenon particularly problematic at slow cooling rates.
Intracellular Ice Formation (Fast Cooling Damage): At rapid cooling rates, water within cells does not have sufficient time to exit before reaching temperatures where ice nucleation occurs spontaneously. This results in the formation of intracellular ice crystals that physically disrupt subcellular organelles, membranes, and the cytoskeleton, typically causing immediate cell death [62]. The probability of intracellular ice formation increases with cooling rate.
The Two-Factor Hypothesis and Optimal Cooling Rate
The two-factor theory provides a conceptual framework for understanding the relationship between cooling rate and cell survival. It posits an optimal cooling rate that minimizes the combined injury from both solute effects and intracellular ice formation, resulting in an inverted U-shaped survival curve when cell viability is plotted against cooling rate [62]. This optimal rate is cell-type specific, influenced by factors including membrane permeability to water, cell surface area to volume ratio, and the presence and type of cryoprotective agents.
Quantitative Data on Cooling Rate Effects
Experimental Evidence from Cell Recovery Studies
Recent investigations have provided quantitative data on the relationship between cooling rates and cell recovery. A 2025 study examining C2C12 mouse myoblasts demonstrated clear cooling-rate-dependent effects on cell viability:
Table 1: Cell Recovery Rates at Different Cooling Rates [64]
| Cooling Rate (°C/min) | Cell Viability (%) | Key Morphological Observations |
|---|---|---|
| 1 °C/min | 65% | Large FCS channels facilitating effective cell accommodation |
| 10 °C/min | 59% | Narrower FCS channels due to finer ice crystals |
| 30 °C/min | 54% | Finest FCS channels limiting cell accommodation |
Statistical analysis of these results (ANOVA, P = 0.034) confirmed a significant difference between conditions, highlighting the critical role of cooling rate in determining post-thaw outcomes [64].
Freeze-Concentrated Solution (FCS) Morphology
The morphology of the freeze-concentrated solution (FCS) – the liquid phase where solutes become concentrated during freezing – has emerged as an important factor in cell preservation outcomes. Research has demonstrated that cooling rate directly influences FCS structure:
- Slow cooling (1°C/min) promotes the formation of relatively large FCS channels due to ice crystal reorientation, providing sufficient volume for cell accommodation [64].
- Rapid cooling (10-30°C/min) produces fine ice crystals and consequently narrower FCS channels, reducing the probability of cell accumulation in these protective regions [64].
The width profile of FCS channels directly influences cell accumulation behavior during freezing, underscoring why cooling rate optimization is essential for designing effective cryopreservation protocols [64].
Methodologies for Cooling Rate Optimization
Experimental Protocols for Cooling Rate Studies
Standardized Freezing-Thawing Protocol for Cell Viability Assessment
A comprehensive approach to evaluating cooling rate effects involves a controlled freezing-thawing process with post-thaw viability assessment:
Sample Preparation: Suspend target cells in cryoprotectant solution (e.g., DMSO at concentrations typically between 5-10% w/v). For morphological studies, addition of fluorescent markers such as sodium fluorescein (100 μM) enables visualization of FCS formation [64].
Cooling Procedure: Aliquot cell suspension (e.g., 10 μL for microscopic studies) into appropriate containers. Cool samples at defined rates (e.g., 1°C/min, 10°C/min, 30°C/min) to an intermediate temperature (e.g., -60°C) using a programmable freezer or controlled cooling device [64].
Terminal Freezing: Transfer samples to ultra-low temperature environment (e.g., -100°C) at a rapid cooling rate (50°C/min) to simulate liquid nitrogen immersion [64].
Thawing Process: Rapidly thaw samples (50°C/min) to minimize ice recrystallization damage [64].
Viability Assessment: Evaluate cell viability using trypan blue exclusion assay or fluorescent viability markers. Calculate viability as the ratio of viable cells to total cells [64].
FCS Morphological Analysis Protocol
To correlate cooling rate with FCS morphology:
Sample Preparation: Prepare DMSO solutions (5, 10, 20 wt%) with sodium fluorescein (100 μM) as fluorescent marker [64].
Microscopic Setup: Utilize an upright fluorescent microscope equipped with a cooling stage and CMOS camera. Sandwich sample (10 μL) between slide glasses on cooling stage [64].
Image Acquisition: Acquire fluorescence images during controlled cooling at various rates. For cell accommodation studies, use rabbit red blood cells dispersed in DMSO [64].
Quantitative Analysis: Measure FCS channel width using ImageJ software. Analyze 40 non-overlapping FCS channels per condition. Determine ice particle size using particle analyzing method in ImageJ [64].
Advanced Cooling Protocols
Interrupted Cooling Protocols
Interrupted cooling strategies offer refined control over the freezing process:
Two-Step Freezing: Initial rapid cooling to a specific sub-zero temperature followed by a holding period before plunging into liquid nitrogen. This approach allows time for intracellular water to exit cells during the hold period, reducing intracellular ice formation during subsequent cooling [65].
Graded Freezing: Systematic cooling to progressively lower temperatures with holds at each stage. This method enables study of how permeating and non-permeating cryoprotectants protect cells differently across temperature ranges [65].
The plunge temperature (transfer to liquid nitrogen) is a critical parameter that significantly impacts cell survival and requires optimization for each cell type [65].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagent Solutions for Cooling Rate Studies
| Item | Function/Application | Example Specifications |
|---|---|---|
| Controlled-Rate Freezer (CRF) | Precisely controls cooling rate during freezing process; enables documentation for cGMP manufacturing [66] | Programmable freezers with temperature monitoring and recording capabilities |
| Dimethyl Sulfoxide (DMSO) | Permeating cryoprotectant; inhibits intracellular ice formation [64] | Typically used at 5-10 wt% concentration; cell culture grade [64] |
| Hydroxyethyl Starch | Non-permeating cryoprotectant; provides extracellular protection [64] | Macromolecular additive; reduces toxic CPA requirements |
| Sodium Fluorescein | Fluorescent marker for FCS visualization during freezing [64] | 100 μM in DMSO solutions for microscopic studies [64] |
| Cell Viability Assays | Quantifies post-thaw cell survival and functionality | Trypan blue exclusion; CCK-8 assay; flow cytometry with viability markers [64] |
| Microscopy System with Cooling Stage | Enables real-time observation of ice formation and FCS morphology | Upright fluorescent microscope with CMOS camera and precision cooling stage [64] |
Current Challenges and Industry Perspectives
Implementation in Cell and Gene Therapy
The cell and gene therapy industry faces significant challenges in cryopreservation optimization. A 2025 survey by the ISCT Cold Chain Management & Logistics Working Group revealed that:
- 87% of respondents use controlled-rate freezing for cell-based products [66]
- 60% utilize default CRF profiles without cell-specific optimization [66]
- 33% dedicate significant resources toward freezing process development [66]
Notably, those experiencing challenges with default profiles typically work with sensitive cell types including iPSCs, hepatocytes, cardiomyocytes, and engineered cells like CAR-T cells [66]. This highlights the need for cell-specific cooling rate optimization, particularly for advanced therapeutic applications.
The Scalability Challenge
Scaling cryopreservation processes represents a major hurdle for the industry, with 22% of survey respondents identifying "Ability to process at a large scale" as the biggest challenge to overcome [66]. This scalability issue complicates cooling rate optimization, as optimal parameters established at research scale may not translate directly to manufacturing-scale processes.
Optimizing cooling rates to balance intracellular ice formation against solute effects remains a fundamental challenge in cryopreservation science. The quantitative relationship between cooling rate and cell viability, coupled with emerging understanding of FCS morphology, provides a scientific foundation for protocol development. As the field advances, particularly in cell and gene therapy, the implementation of robust, standardized methodologies for cooling rate optimization will be essential for ensuring consistent post-thaw cell viability and functionality. Future directions will likely include increased automation, advanced modeling of heat and mass transfer, and development of novel cryoprotectant formulations that widen the optimal cooling rate window for sensitive cell types.
This technical guide explores two advanced technologies—Magnetic Field (MF)-Assisted Freezing and Controlled Nucleation—within the critical context of managing the mechanical and osmotic stresses on cells during freezing. The freezing of biological materials, from therapeutic cells to food matrices, subjects them to two primary insults: the mechanical damage from ice crystal formation and the osmotic stress resulting from solute concentration in the unfrozen fraction. This whitepaper details how these novel approaches directly counter these cryo-injuries, providing researchers and drug development professionals with the scientific principles, experimental protocols, and practical data needed for implementation.
Core Principles and Cellular Context
The Mechanical and Osmotic Effects of Freezing on Cells
During slow cooling, ice formation begins in the extracellular space. This sequesters pure water as ice, thereby concentrating the solutes in the remaining unfrozen extracellular fluid. The resulting osmotic imbalance causes water to osmotically exit the cell, leading to intracellular dehydration and cell shrinkage. This excessive dehydration causes mechanical stress on the cell membrane and can lead to a loss of membrane integrity, a phenomenon identified as a primary cause of cryo-injury in slow cooling [67]. Conversely, if cooling is too rapid, intracellular water does not have sufficient time to exit the cell. Consequently, the cell interior becomes supercooled, leading to intracellular ice formation (IIF), which is almost always lethal to the cell [68]. Therefore, the fundamental challenge in cryopreservation is to manage the trade-off between dehydration and ice formation.
The Role of the Investigated Technologies
The two technologies addressed herein offer distinct pathways to mitigate these damaging effects:
- Controlled Nucleation: This technique directly manages the ice formation process itself. By precisely initiating ice formation (nucleation) at a defined, higher temperature, it reduces the degree of supercooling. This promotes the formation of larger, more regular extracellular ice crystals and provides more time for controlled water efflux from cells, thereby minimizing intracellular ice formation and reducing damaging mechanical stresses [68] [69].
- Magnetic Field (MF)-Assisted Freezing: This approach focuses on stabilizing the cell membrane against osmotic stress. Exposure to a static magnetic field (SMF) has been demonstrated to decrease membrane fluidity by aligning diamagnetic phospholipid molecules, thereby increasing membrane rigidity and stability. A more stable membrane is better able to resist the mechanical stresses of dehydration and volume change during slow cooling [67] [70].
Magnetic Field-Assisted Freezing
Mechanism of Action
The primary cryoprotective mechanism of SMFs is the stabilization of the cell membrane. The major composite component of cell membranes, phospholipid, is a molecule that exhibits a highly diamagnetic anisotropic susceptibility. When exposed to an SMF, phospholipids experience a torque force that orients them, thereby increasing membrane rigidity and decreasing membrane fluidity [67]. This effect was quantitatively demonstrated in human erythrocytes, where exposure to a 0.8-T SMF significantly decreased membrane fluidity in the hydrophobic regions [67]. This enhanced membrane stability allows cells to better resist the dehydration damage and mechanical stresses caused by slow cooling procedures [67] [70]. Furthermore, MF exposure is also reported to strengthen the supercooling state, promote nucleation, and prevent the formation of large, damaging ice crystals, thereby helping to preserve cellular structure [70].
Experimental Protocol: SMF Exposure during Erythrocyte Slow Freezing
The following protocol is adapted from a study investigating the cryoprotective effect of SMFs on human erythrocytes frozen in a low concentration of glycerol [67].
1. Sample Preparation:
- Material: Obtain human erythrocytes from whole blood. Wash and centrifuge (e.g., 330× g for 14 min) to remove plasma and buffy coat. Adjust the packed erythrocytes to a hematocrit of 75% (v/v) in phosphate-buffered saline (PBS).
- Cryoprotectant: Glycerolize the samples by slowly mixing the erythrocyte suspension with a 35% (w/v) glycerol solution to achieve a final glycerol concentration of 20% (w/v). This sub-optimal glycerol concentration is used to sensitize the system to the SMF effect.
- Loading: Load 0.5 mL of the glycerolized sample into 0.6-mL tubes.
2. SMF Exposure and Freezing:
- Apparatus: Place sample tubes in a custom-built sample chamber equipped with NdFeB permanent magnets and iron yokes to generate a uniform SMF. For controls, use an identical setup with non-magnetized NdFeB blocks (0 T).
- Field Strength: Expose samples to SMF strengths of 0.4 T or 0.8 T.
- Freezing Program: Use a two-step cooling program in a computer-controlled freezer.
- Hold at -5°C for 10 minutes.
- Cool from -5°C to -55°C at a controlled rate of 1°C/min.
- Storage: Immediately transfer frozen samples to a -80°C mechanical freezer (without an SMF) for 24 hours.
3. Thawing and Assessment:
- Thawing: Rapidly thaw samples in a 37°C water bath for 1 minute.
- Viability Assessment:
- Survival Rate: Perform a hemolysis test using Drabkin's reagent to measure total and supernatant hemoglobin. Calculate the survival rate and normalize data to the control to report the relative survival ratio.
- Morphology: Observe thawed erythrocytes using an optical microscope and analyze the mean corpuscular volume (MCV) with an automatic cell analyzer.
- Membrane Properties: Assess membrane fluidity using fluorescence anisotropy probes and conduct dehydration stability tests.
Key Experimental Data and Outcomes
Table 1: Summary of Key Findings from Erythrocyte SMF Freezing Study [67]
| Parameter | Control (0 T) | 0.4 T SMF | 0.8 T SMF | Measurement Method |
|---|---|---|---|---|
| Relative Survival Ratio | 1.0 (Baseline) | Increased by ~10% | Increased by ~20% (p<0.001) | Hemolysis test / Drabkin's reagent |
| Membrane Fluidity | Baseline | Not Reported | Significant decrease (p<0.05) in hydrophobic regions | Fluorescence anisotropy |
| Dehydration Stability | Baseline | Not Reported | Significantly lower hemolysis (p<0.05) | Osmotic challenge assay |
| Morphology & MCV | No significant changes observed | Optical microscopy, cell analyzer |
Controlled Nucleation in Freezing and Freeze-Drying
Mechanism of Action
Controlled nucleation directly addresses the stochastic nature of ice formation. In uncontrolled freezing, ice nucleates randomly in time and temperature (e.g., between -5°C and -15°C), leading to high variability in ice crystal size and morphology within a batch [69]. Controlled nucleation allows the user to assign both the time and temperature at which nucleation occurs, a concept known as "two-dimensional control" [69]. By initiating nucleation at a higher, defined temperature (e.g., -3°C to -6°C), the degree of supercooling is reduced. This results in the formation of larger and more uniform ice crystals. Larger crystals create larger pores in the dried product matrix (if lyophilized), which drastically reduces the resistance to water vapor flow (Rp) during primary drying. This leads to faster drying rates, shorter process times, and improved batch homogeneity [69]. In cell cryopreservation, controlled nucleation at a higher temperature (e.g., -6°C) has been shown to promote more intracellular dehydration and less intracellular ice formation, correlating with higher post-thaw viability in T cells [68].
Technical Methodologies for Inducing Nucleation
Several technical concepts have been developed to implement controlled nucleation:
- Ice Fog Technique: Cold nitrogen gas is introduced into the chamber, causing moisture to freeze and form a dense "ice fog." These tiny ice crystals settle on the surface of the product solutions, inducing nucleation. Modern implementations use a steam generator and heat exchanger to ensure a uniform fog in large-scale equipment [69].
- Depressurization (Vacuum-Induced Surface Freezing): The product solution is first equilibrated on a cooled shelf at a temperature slightly below its equilibrium freezing point. The chamber is then briefly pressurized with an inert gas (e.g., argon to ~2.94 bar), after which the overpressure is rapidly released (within ~10 seconds). The sudden pressure drop causes adiabatic cooling and/or gas bubble formation, which induces nucleation from the top of the vials downward [69].
- Seeding: A small amount of crystalline material of the solute is added to the solution to act as nucleation sites. While it increases the nucleation temperature, it does not offer precise "two-dimensional control" over the nucleation event [69].
- Ultrasound Nucleation: Applying a short acoustic pulse (10-40 kHz) to the supercooled solution induces cavitation, which effectively initiates nucleation. This method can produce many small crystals with a narrow size distribution but has seen limited large-scale adoption [71] [69].
Experimental Protocol: Controlled Nucleation via Depressurization for Freeze-Drying
This protocol outlines the steps for implementing controlled nucleation in a freeze-drying cycle using the depressurization method [69].
1. Pre-freezing and Equilibration:
- Load the product vials onto the freeze-dryer shelf.
- Cool the shelf to a target temperature slightly below the product's equilibrium freezing point (Tf). For a sucrose model formulation, this could be around -3°C.
- Hold the shelf at this temperature to allow the product to equilibrate. Ensure the product is supercooled and has not yet nucleated.
2. Depressurization Nucleation Cycle:
- Pressurization: Isolate the chamber from the vacuum pump and introduce an inert gas (e.g., sterile argon or nitrogen) to pressurize the chamber to a predefined level, typically around 2.8-2.9 bar absolute (~28 psig).
- Hold: Maintain the pressure for a short period (e.g., 10-30 seconds).
- Rapid Depressurization: Quickly vent the chamber pressure back to atmospheric levels or to the target primary drying pressure, ideally within 10 seconds or less. This rapid pressure drop will induce nucleation across the entire batch simultaneously.
- Observation: Nucleation will typically be observed progressing from the top of the vial downwards, contrary to conventional shelf freezing.
3. Freezing and Drying:
- Immediately after nucleation, lower the shelf temperature to fully solidify the product (e.g., to -45°C).
- Once freezing is complete, initiate primary drying by applying a vacuum and controlling the shelf temperature to sublime the ice.
4. Process Monitoring:
- Compare the resistance of the dried product layer (Rp) and primary drying times against a batch frozen with uncontrolled nucleation. Controlled nucleation at a higher temperature should result in a significantly lower Rp and shorter primary drying time [69].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Materials for Implementing the Described Techniques
| Item | Function / Application | Example / Specification |
|---|---|---|
| Static Magnetic Field Setup | Generates a uniform magnetic field for MF-assisted freezing experiments. | NdFeB (N52) permanent magnets with iron yokes; Gauss meter for flux density measurement (e.g., 0.4 T, 0.8 T) [67]. |
| Cryoprotectants | Protect cells from dehydration and ice damage during slow freezing. | Glycerine (20% w/v for erythrocyte model) [67]; Dimethyl Sulfoxide (DMSO, e.g., 2.5-5% v/v for T cells) [68]. |
| Controlled Nucleation Equipment | Enables precise initiation of ice formation in freeze-drying. | Freeze-dryer with depressurization capability (e.g., SMART Freeze Dryer with Nucleation on-Demand) [69]. |
| Osmotic Solution | Acts as the medium for osmotic dehydration and compound infusion. | Pomegranate juice concentrate (45-65° Brix) for studying osmotic mass transfer [72]. |
| Cell Culture Materials | Model systems for studying cellular cryo-injury. | Human erythrocytes [67]; Jurkat T cells [68]. |
| Viability Assays | Quantify post-thaw survival and membrane integrity. | Hemolysis test with Drabkin's reagent [67]; Membrane integrity assay (e.g., propidium iodide exclusion) [68]. |
Integrated Workflow and Conceptual Diagrams
The following workflow and diagram synthesize the core concepts and experimental paths discussed in this guide.
Experimental Workflow for Investigating Novel Freezing Approaches
Cellular Response to Freezing Pathways
This diagram illustrates the different cellular outcomes resulting from conventional versus novel freezing approaches.
The integration of Magnetic Field-Assisted Freezing and Controlled Nucleation represents a significant advancement in the control of freezing processes. By directly targeting the fundamental mechanical and osmotic causes of cryo-injury—through membrane stabilization and precise management of ice formation, respectively—these technologies offer researchers powerful tools to improve the post-preservation viability of a wide range of biological materials. The experimental protocols and data summarized in this guide provide a foundation for the further development and application of these novel approaches in pharmaceutical development and biopreservation.
Application-Specific Protocols for Mammalian Cells, Tissues, and Probiotics
Cryopreservation is a fundamental technique for preserving biological materials, yet the process subjects cells to severe mechanical and osmotic stresses that can compromise their viability and functionality. The core challenge lies in managing the phase change of water, which triggers two primary damage mechanisms: intracellular ice formation causing mechanical damage to cellular structures, and osmotic dehydration leading to solute concentration effects and cell shrinkage [5]. Successful cryopreservation protocols must therefore balance these competing injuries through optimized cooling rates and cryoprotective agents.
The mechanical effects manifest predominantly through ice crystal formation. During slow freezing, extracellular ice formation initiates, creating a solute gradient that draws water out of cells, causing protective dehydration but potentially leading to excessive volumetric shrinkage and "solution effects" from concentrated electrolytes [73]. Conversely, rapid cooling may not allow sufficient time for cellular dehydration, resulting in lethal intracellular ice formation that mechanically disrupts organelles and membrane systems [5]. For probiotics, these freezing injuries occur primarily during the freezing stage, with studies showing significantly greater viability loss during drying than during freezing itself [74].
This technical guide examines application-specific protocols for mammalian cells, tissues, and probiotics within this mechanistic framework of freezing injury, providing researchers with evidence-based methodologies to maximize post-preservation recovery.
Mammalian Cell Cryopreservation Protocols
Fundamental Freezing Principles for Mammalian Cells
The foundational principle for mammalian cell cryopreservation involves slow freezing and rapid thawing. Slow freezing at approximately -1°C/minute facilitates protective cellular dehydration, while rapid thawing minimizes damaging ice recrystallization [75]. For sensitive cell types like induced pluripotent stem cells (iPSCs), maintaining a cooling rate between -0.3°C/min to -1.8°C/min is critical for optimal survival [5]. These parameters must be carefully optimized as cells are particularly vulnerable to intracellular ice formation, which mechanically damages cell membranes [5].
Cryoprotectant agents function primarily by modulating osmotic responses during freezing. Dimethyl sulfoxide (DMSO), the most common cryoprotectant, creates a hypertonic environment (approximately 1.4 osm/L for 10% DMSO solutions) that promotes controlled cellular dehydration while penetrating cells to prevent intracellular ice formation [5]. The equilibrium between extracellular ice formation and cellular dehydration represents the delicate balance necessary for successful cryopreservation.
Standardized Mammalian Cell Freezing Protocol
The following protocol applies to most adherent and suspension mammalian cell cultures, with specific considerations for different cell types detailed in subsequent sections [7]:
- Pre-freezing Preparation: Harvest cells during log-phase growth at >80% confluency with >90% viability. Characterize cells and confirm absence of microbial contamination before freezing [7] [75].
- Cryoprotectant Medium Preparation: Prepare freezing medium consisting of complete growth medium supplemented with 10% DMSO or a specialized commercial cryopreservation medium. Maintain at 2°-8°C until use [7].
- Cell Detachment and Resuspension: For adherent cells, gently detach using appropriate dissociation reagent. Resuspend cells in complete growth medium and determine viable cell density using Trypan Blue exclusion or automated cell counting [7].
- Centrifugation and Cryomedium Addition: Centrifuge cell suspension at 100-400 × g for 5-10 minutes. Aspirate supernatant and resuspend cell pellet in cold freezing medium at recommended density (typically 1×10^6 to 1×10^7 cells/mL) [7] [75].
- Aliquoting and Controlled-Rate Freezing: Dispense cell suspension into sterile cryogenic vials. Freeze cells using a controlled-rate freezer or isopropanol freezing container placed at -80°C overnight to achieve approximate cooling rate of -1°C/minute [7] [75].
- Long-Term Storage: Transfer frozen cryovials to liquid nitrogen storage (-135°C to -196°C) in the vapor phase to prevent potential explosive hazards associated with liquid phase storage [7].
Table 1: Optimal Freezing Parameters for Specific Mammalian Cell Types
| Cell Type | Freezing Medium | Cell Concentration | Cooling Rate | Special Considerations |
|---|---|---|---|---|
| Standard Cell Lines | Complete medium + 10% DMSO | 1×10^6 - 5×10^6 cells/mL | -1°C/min | Freeze at high viability during log-phase growth [7] |
| iPSCs | mFreSR or CryoStor CS10 | As cell aggregates | -1°C to -3°C/min | Passage as aggregates preserves cell-cell contacts; avoid single-cell freezing when possible [5] |
| Mesenchymal Stem Cells | MesenCult-ACF Freezing Medium | 1×10^6 - 5×10^6 cells/mL | -1°C/min | Use specialized media for maintained differentiation potential [75] |
| PBMCs | CryoStor CS10 or FBS + 10% DMSO | 5×10^6 - 1×10^7 cells/mL | -1°C/min | Consider laboratory-specific optimization for recovery [75] |
| hPSC-Derived Cardiomyocytes | STEMdiff Cardiomyocyte Freezing Medium | Manufacturer recommended | -1°C/min | Use lineage-specific media for optimal functional recovery [75] |
Specialized Considerations for iPSCs and Stem Cells
Induced pluripotent stem cells require specialized handling to maintain pluripotency and high viability after thawing. These sensitive cells exhibit particular vulnerability to intracellular ice formation, necessitating precise control over cooling rates [5]. Research indicates that a three-zone temperature profile optimizes iPSC survival: rapid cooling in the dehydration zone, slow cooling in the nucleation zone (-0.3°C/min to -1.8°C/min), and rapid cooling in the final zone [5].
The method of passaging significantly impacts post-thaw recovery. Freezing iPSCs as cell aggregates rather than single cells preserves cell-cell contacts that support survival, though variable aggregate size may cause inconsistent cryoprotectant penetration [5]. Alternatively, single-cell freezing allows better quantification and consistency but requires optimized recovery conditions to reform aggregates.
Probiotic Preservation Methodologies
Freeze-Drying Techniques for Probiotics
Probiotic preservation employs freeze-drying (lyophilization) to achieve long-term stability, with the freezing phase representing the most critical stage for maintaining viability. Research demonstrates that 60-70% of cells that survive the freezing phase will subsequently survive the dehydration process [76]. The fundamental challenge lies in managing ice crystal formation that can rupture cell membranes during slow freezing [76].
Innovative approaches like flash freeze-drying (FFD) have shown significant improvements over conventional freeze-drying. FFD employs cyclic pressure variation (0.4-1000 millibars) during primary drying, reducing total process time by 68.75% (900 min versus 2880 min) while improving outcomes [76]. When treating Lactobacillus acidophilus LA5 at -25°C, FFD achieved cell viability of 89.94% - 2.74% higher than conventional freeze-drying - along with 55% significantly lower water activity (0.0522) [76].
Supercooling pretreatment represents another advanced technique that inhibits ice nucleation to enhance bacterial viability. This approach aims to minimize the mechanical damage caused by ice crystal formation, which currently results in approximately 25% cell viability loss in commercial facilities during standard temperature drops from 37°C to -80°C [77].
Table 2: Optimization of Freezing Parameters for Probiotic Strains
| Freezing Parameter | Conventional Freeze-Drying | Flash Freeze-Drying (FFD) | Supercooling Pretreatment |
|---|---|---|---|
| Process Time | 2880 min (48 h) [76] | 900 min (68.75% reduction) [76] | Research phase |
| Temperature Parameters | -40°C initial freezing [76] | -25°C, -15°C, or -3°C strategies [76] | Sub-zero without nucleation |
| Cell Viability Results | 87.2% for L. acidophilus LA5 [76] | 89.94% at -25°C [76] | Target: >90% (research) [77] |
| Water Activity (a_w) | ~0.116 [76] | 0.0522 (55% improvement) [76] | Not specified |
| Storage Stability | 28 days at 20°C [76] | 64.72% viability after 28 days at 20°C [76] | Improved shelf life projected |
| Key Mechanism | Slow freezing with sublimation | Pressure cycling with flash sublimation | Inhibition of ice nucleation |
Microencapsulation for Enhanced Probiotic Survival
Microencapsulation techniques provide physical protection for probiotic cells during freezing and storage. This approach entraps cells within a protective matrix, typically alginate and chitosan, creating a microenvironment that shields against freezing-induced damage [76]. The encapsulation yield (EY) formula quantifies this protective efficacy:
Where N represents viable cells released from microcapsules and N₀ represents viable cells prior to microencapsulation [76]. This physical barrier minimizes direct ice crystal contact with cell membranes and reduces osmotic shock during the freezing process.
Microencapsulation demonstrates particular value during gastrointestinal transit, with enteric-coated capsules providing gastric protection while minimizing viability losses. Studies with Eudragit L100-55 coated capsules showed about 95% recovery of viable cells after the coating process, with capsules resisting simulated gastric fluid while disintegrating in simulated intestinal fluid [74].
Ice Crystal Formation and Its Mechanical Impact
Freezing Rate Determines Ice Crystal Morphology
The freezing rate fundamentally controls ice crystal size and distribution, directly determining the mechanical damage inflicted on cellular structures. Rapid freezing produces numerous small, uniform ice crystals that minimally disrupt cellular architecture, while slow freezing generates large, irregular ice crystals that cause severe structural damage [78]. In plant tissues like strawberries, higher freezing rates (10.43 cm/h) better preserve original cell structure, while the slowest freezing rates cause the most significant damage due to extensive ice crystal formation [78].
The maximum ice crystal formation zone (-1°C to -5°C) represents the critical temperature range where most tissue ice formation occurs [79]. During this phase transition, free water and immobilized water progressively crystallize while bound water remains molecularly stable. The size distribution and spatial arrangement of ice crystals governed by processing parameters ultimately determine the structural integrity of frozen biological materials [79].
Tissue-Level Mechanical Effects
At the tissue level, freezing induces spatiotemporal redistribution of interstitial fluid and subsequent extracellular matrix (ECM) swelling that contributes to microstructural changes [17]. Engineered tissue studies using cell image deformetry (CID) have quantified these freezing-induced deformations, revealing that cell-matrix interactions provide mechanical support to minimize expansion regions during freezing [17].
The dilatation (volumetric strain) in tissues during freezing can be quantified using the formula:
Where u and v represent deformation rates in the x and y directions respectively [17]. This mechanical strain results from the complex interplay between cellular water transport and extracellular ice formation, with cell-ECM adhesion significantly influencing the resulting deformation patterns.
Osmotic Stress During Freezing
Osmotic Damage Mechanisms
Osmotic stress represents the second major mechanism of freezing injury, complementing the mechanical damage caused by ice crystals. As extracellular ice forms, solute concentration in the unfrozen fraction increases, creating osmotic pressure gradients that draw water out of cells [79] [73]. This cellular dehydration concentrates intracellular solutes to potentially toxic levels and causes volumetric shrinkage that may exceed tolerable limits [73].
The interplay between cooling rate and osmotic response creates a critical balance. Slow cooling permits extensive cellular dehydration but risks "solution effects" from concentrated electrolytes, while rapid cooling limits dehydration but promotes intracellular ice formation [5]. This fundamental relationship explains why optimal cooling rates are cell type-specific, depending on membrane permeability characteristics and surface-to-volume ratios [5].
Cryoprotectant Mechanisms Against Osmotic Stress
Cryoprotectants function primarily as osmotic regulators during freezing. Permeating cryoprotectants like DMSO enter cells and reduce the electrolyte concentration gradient that drives dehydration, while simultaneously reducing the amount of water converted to ice at any given temperature [5]. Non-permeating cryoprotectants like hydroxyethyl starch create extracellular osmotic forces that control dehydration rate and extent.
The optimal introduction of cryoprotectants requires careful temperature management. At room temperature, cells placed in cryoprotectant solutions experience initial osmotic shrinkage as water exits rapidly, followed by return to original volume as the cryoprotectant permeates [5]. This osmotic response varies significantly between cell types, requiring protocol optimization for different biological systems.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Cryopreservation Research
| Reagent Category | Specific Examples | Function & Mechanism | Application Notes |
|---|---|---|---|
| Cryoprotectant Agents | DMSO, Glycerol, Ethylene glycol | Reduce freezing point, slow cooling rate, prevent intracellular ice formation [7] | DMSO at 10% concentration standard for many cell types; use culture-grade, open only in laminar flow hood [7] |
| Specialized Freezing Media | CryoStor CS10, mFreSR, Synth-a-Freeze | Chemically-defined formulations with optimized cryoprotectant ratios [7] [75] | CryoStor CS10 for general use; mFreSR for iPSCs; protein-free options available for sensitive applications [75] |
| Cell Detachment Reagents | Trypsin, TrypLE Express, Accutase | Gently detach adherent cells with minimal damage to surface proteins [7] | Phenol-red free formulations recommended to avoid interference with downstream analysis [7] |
| Viability Assessment | Trypan Blue, Automated cell counters (Countess) | Determine viable cell density and percent viability before freezing [7] | Critical quality control step; aim for >90% viability pre-freezing [7] |
| Cryogenic Containers | Mr. Frosty, CoolCell | Achieve controlled-rate freezing at approximately -1°C/minute in standard -80°C freezers [7] [75] | Isopropanol-containing or isopropanol-free options available; provide consistent cooling rates without specialized equipment [75] |
| Storage Vessels | Sterile cryogenic vials, Liquid nitrogen tanks | Maintain long-term stability below glass transition temperatures [7] [5] | Internal-threaded vials prevent contamination; vapor phase storage reduces explosion risks [7] [75] |
| Microencapsulation Materials | Sodium alginate, Chitosan, Calcium chloride | Form protective matrix around probiotic cells [76] | Creates physical barrier against ice crystal damage; improves gastrointestinal survival for probiotics [76] |
The mechanical and osmotic stresses imposed during freezing present complex challenges that require application-specific solutions across biological systems. For mammalian cells, controlled-rate freezing with optimized cryoprotectant formulations balances dehydration and intracellular ice formation, with specialized approaches needed for sensitive types like iPSCs. For probiotics, techniques like flash freeze-drying and microencapsulation address the particular vulnerability of bacterial membranes to ice crystal damage. Understanding these fundamental mechanisms of freezing injury enables researchers to select appropriate parameters and protective strategies for their specific biological materials, ultimately enhancing post-preservation viability and functionality.
Optimizing Cryopreservation Outcomes: Strategies to Mitigate Freezing Damage and Enhance Cell Recovery
Cooling Rate Optimization for Different Cell Types and Sizes
The process of cryopreservation is a cornerstone of modern biological research and cell-based therapies, enabling long-term storage of living cells by suspending their metabolic activities at ultra-low temperatures. The cooling rate during freezing stands as one of the most critical parameters determining post-thaw cell viability and functionality. Within the broader context of research on mechanical and osmotic effects of freezing on cells, cooling rate optimization directly addresses the fundamental challenge of balancing two competing injury mechanisms: intracellular ice formation (a mechanical effect) and cellular dehydration (an osmotic effect).
When cells are cooled below freezing temperatures, water initially freezes extracellularly, creating a hypertonic environment that draws water out of cells through osmosis. The cooling rate determines whether this water transport can occur efficiently. Slow cooling permits extensive cellular dehydration, minimizing intracellular ice formation but potentially causing damaging solute concentration effects and excessive cell shrinkage. In contrast, rapid cooling does not allow sufficient time for water efflux, resulting in supercooling and lethal intracellular ice formation when water eventually freezes within the cell [80]. This delicate balance varies significantly across different cell types and sizes due to fundamental differences in their membrane permeability properties and surface-to-volume ratios [65].
The optimization of cooling protocols is particularly crucial for the advancing field of cell-based therapies, where preserving cell viability, potency, and functionality after thawing is essential for therapeutic efficacy. Different cell types exhibit markedly different optimal cooling rates based on their biological characteristics, necessitating customized approaches to cryopreservation [81]. This technical guide provides a comprehensive framework for optimizing cooling rates across diverse cell types, with specific methodologies and data-driven recommendations for researchers and drug development professionals.
Fundamental Principles of Freezing Injury
Mechanical and Osmotic Injury Mechanisms
The freezing of cellular suspensions subjects cells to two primary classes of injury: mechanical damage from ice crystals and osmotic stress from solute concentration gradients. Understanding these interrelated mechanisms is essential for developing optimized cryopreservation protocols.
Mechanical injury from intracellular ice formation (IIF) is typically lethal to cells. IIF occurs when the cooling rate is too rapid to permit sufficient cellular dehydration, resulting in the nucleation and growth of ice crystals within the cytoplasm. These crystals can physically disrupt organelles, rupture membranes, and damage the cytoskeleton [80]. The probability of IIF increases with cooling rate and is influenced by cell-specific factors including membrane permeability and surface area to volume ratio [13].
Osmotic injury occurs during slower freezing processes. As extracellular ice forms, solutes are excluded from the growing ice lattice, creating a hypertonic extracellular environment. This osmotic imbalance draws water out of cells, leading to cellular dehydration and shrinkage. Excessive dehydration can cause concentration of intracellular solutes to toxic levels, membrane damage from lipid restructuring, and reduction of cell volume below a critical minimum [80] [65]. The extent of dehydration is inversely related to cooling rate – slower cooling permits more complete dehydration but increases exposure to concentrated solutes.
The relationship between cooling rate and cell survival typically follows an "inverted U" curve, where survival is maximal at an optimal cooling rate that balances these competing injury mechanisms. At cooling rates below this optimum, cells experience excessive dehydration damage, while rates above the optimum promote lethal intracellular ice formation [65].
The Impact of Cell Size and Type on Freezing Response
Cell-specific characteristics significantly influence their response to freezing and thus their optimal cooling rate. The most important factors include:
Surface-to-Volume Ratio: This geometric property determines the rate at which water can exit a cell in response to osmotic gradients. Smaller cells with high surface-to-volume ratios can dehydrate more rapidly than larger cells, making them more tolerant to faster cooling rates [65].
Membrane Permeability to Water: The hydraulic conductivity (Lp) of the cell membrane governs water transport during freezing. Cells with higher membrane permeability dehydrate more efficiently and typically tolerate faster cooling rates. Membrane permeability is both cell-type dependent and temperature-dependent, decreasing significantly as temperatures decline [13].
Cell Function and Architecture: Complex cells with extensive processes or specialized organelles may have particular vulnerabilities to freezing injury. For example, neurons with long axons and dendritic networks are particularly susceptible to ice crystal damage [81].
The following diagram illustrates how these competing injury mechanisms and cellular characteristics interact during the freezing process:
Cooling Rate Optimization by Cell Type
Quantitative Recommendations for Different Cell Types
Optimal cooling rates vary significantly across cell types due to differences in membrane permeability, size, and biological structure. The following table summarizes evidence-based cooling rate recommendations for common cell types used in research and therapy:
Table 1: Optimal Cooling Rate Recommendations by Cell Type
| Cell Type | Optimal Cooling Rate Range | Key Considerations | Supporting Evidence |
|---|---|---|---|
| T-cells & Lymphocytes | -1°C/min to -3°C/min | Controlled-rate freezing preferred for consistent results | [66] [81] |
| hPSCs (iPSCs/ESCs) | -1°C/min | Highly sensitive to intracellular ice; precise control critical | [5] [75] |
| Oocytes | -0.3°C/min to -50°C/min (two-step) | Large size increases susceptibility to ice formation | [5] |
| MSCs | -1°C/min to -2°C/min | Moderate sensitivity; consistent protocols needed for therapeutics | [75] [81] |
| Neurons | -1°C/min to -2°C/min | Complex architecture increases vulnerability to ice damage | [81] |
| Hepatocytes | -1°C/min to -3°C/min | Require optimized protocols for differentiated function | [81] |
| Red Blood Cells | -100°C/min to -1000°C/min | Very high membrane permeability to water | [65] |
Advanced Optimization Strategies
Interrupted Cooling Protocols
Sophisticated cooling strategies that incorporate precisely timed rate changes or temperature holds can significantly improve cell survival compared to linear cooling protocols. These "interrupted cooling" approaches actively manipulate the balance between dehydration and intracellular ice formation:
Two-Step Freezing: This approach involves an initial rapid cooling step to an intermediate sub-zero temperature (typically between -15°C and -40°C), followed by a holding period to allow cellular dehydration, before final rapid cooling to cryogenic temperatures. This method is particularly beneficial for cell types with moderate membrane permeability [65].
Three-Zone Optimization: Research on human induced pluripotent stem cells (iPSCs) suggests that a "fast-slow-fast" cooling pattern optimizes survival: rapid cooling through the dehydration zone, slow cooling through the intracellular ice formation (nucleation) zone, and rapid cooling again at lower temperatures [5].
The following diagram illustrates this sophisticated three-zone optimization approach:
Scaling Considerations for Cell Therapy
The transition from research-scale to therapeutic-scale cryopreservation presents unique challenges for cooling rate optimization. Controlled-rate freezers (CRFs) provide precise control over cooling parameters and are considered essential for manufacturing cell therapies, particularly at later clinical stages [66]. However, several considerations emerge at scale:
Profile Optimization: While 60% of surveyed organizations use default CRF profiles, many sensitive cell types (including iPSCs, hepatocytes, cardiomyocytes, and neural cells) require customized cooling profiles for optimal recovery [66].
Container Effects: The cooling rate experienced by cells is influenced by container type, volume, and load configuration within the CRF. Qualification studies should include temperature mapping across different container types and locations [66].
Process Consistency: For cell therapies, cryopreserving entire manufacturing batches together minimizes inter-batch variability, though this practice presents scaling challenges for large production volumes [66].
Experimental Protocols for Cooling Rate Optimization
Standard Controlled-Rate Freezing Protocol
For researchers establishing cooling rate optimization experiments, the following protocol provides a foundation for systematic evaluation:
Cell Preparation:
Cryoprotectant Addition:
Sample Aliquotting:
- Dispense cell suspension into sterile cryogenic vials
- Maintain consistent fill volumes across samples for comparable cooling kinetics
- Use internal-threaded cryovials to prevent contamination during storage [75]
Controlled-Rate Freezing:
Cryogenic Storage:
Methodology for Cooling Rate Determination
To empirically determine optimal cooling rates for novel cell types:
Experimental Design:
- Test a range of cooling rates (e.g., -0.1°C/min, -0.5°C/min, -1°C/min, -5°C/min, -10°C/min, -50°C/min)
- Include sufficient replicates for statistical power (n≥3)
- Use consistent cell concentration across conditions
Viability Assessment:
- Employ multiple assessment methods 24 hours post-thaw:
- Membrane integrity: Trypan blue exclusion
- Metabolic activity: MTT, PrestoBlue, or ATP assays
- Apoptosis markers: Annexin V staining
- Lineage-specific functionality assays [80]
- Employ multiple assessment methods 24 hours post-thaw:
Data Analysis:
- Plot post-thaw viability versus cooling rate to identify optimum
- Compare outcomes across different assessment methods
- Evaluate morphology and attachment for adherent cells
The Scientist's Toolkit: Essential Reagents and Equipment
Table 2: Essential Materials for Cooling Rate Optimization Research
| Category | Specific Products/Solutions | Function & Application |
|---|---|---|
| Cryoprotectants | DMSO, Glycerol, Ethylene glycol, Commercial media (CryoStor, mFreSR) | Reduce ice formation; protect from dehydration and mechanical damage |
| Cell Culture Consumables | Cryogenic vials, Serological pipettes, Centrifuge tubes | Maintain sterility; enable precise aliquotting |
| Cooling Rate Control | Controlled-rate freezers, Passive cooling containers (Mr. Frosty, CoolCell) | Implement precise cooling profiles; ensure reproducibility |
| Viability Assessment | Trypan blue, Automated cell counters, Metabolic assays, Flow cytometry reagents | Quantify post-thaw recovery; assess multiple viability parameters |
| Storage Systems | Liquid nitrogen tanks, -150°C mechanical freezers | Maintain stable cryogenic temperatures; prevent recrystallization |
Optimizing cooling rates for specific cell types and sizes remains a critical challenge in cryobiology, directly addressing the fundamental balance between mechanical ice injury and osmotic stress during freezing. The empirical data and methodologies presented in this guide provide a framework for developing cell-type specific cryopreservation protocols that maximize post-thaw viability and functionality.
As cell therapies continue to advance toward clinical application, precise control over cooling parameters will become increasingly important for manufacturing consistency and therapeutic efficacy. Future directions in the field include the development of DMSO-free cryopreservation methods that maintain cell viability while eliminating concerns about cryoprotectant toxicity, particularly for novel administration routes in cell therapy. Additionally, more sophisticated interrupted cooling protocols that dynamically respond to the changing biophysical needs of cells during freezing represent promising approaches for further enhancing cryopreservation outcomes across diverse cell types.
Cryopreservation serves as a cornerstone technology for maintaining the viability and functionality of biological systems across pharmaceutical development, biomedical research, and clinical applications. The fundamental challenge lies in mitigating the lethal mechanical and osmotic effects of freezing on cellular structures. During freezing, ice formation externally concentrates solutes, creating osmotic stress that dehydrates and shrinks cells, while intracellular ice crystals can mechanically disrupt membranes and organelles [2] [42]. Cryoprotectant Agents (CPAs) are specifically formulated compounds designed to counteract these damaging processes, enabling long-term storage of cells, tissues, and increasingly complex biological constructs at cryogenic temperatures.
The formulation of effective cryoprotectant solutions requires a delicate balance between achieving sufficient protection and minimizing CPA-induced toxicity. As research advances toward preserving more complex systems like tissue-engineered constructs and organs, this balance becomes increasingly critical [82] [52]. This technical guide examines current cryoprotectant formulation strategies within the context of mechanical and osmotic freezing damage, providing researchers with evidence-based approaches for optimizing preservation protocols across diverse biological applications.
Mechanisms of Freezing Damage and Cryoprotectant Action
Primary Mechanisms of Freezing Damage
The transition of water to ice during cooling triggers multiple interdependent damage pathways:
- Solution Effects: As extracellular ice forms, dissolved solutes become concentrated in the remaining liquid phase, creating hypertonic conditions that osmotically dehydrate cells. This elevated solute concentration can denature proteins, disrupt lipid membranes, and cause metabolic dysfunction [42].
- Intracellular Ice Formation (IIF): With rapid cooling, insufficient time for osmotic water efflux leads to supercooling and eventual intracellular freezing. IIF is almost universally lethal, causing mechanical damage to membranes, organelles, and cytoskeletal structures [2] [42].
- Membrane Phase Transitions: Dehydration and cold temperatures can induce lipid membrane transitions from fluid lamellar phases to gel-phase or non-lamellar structures, compromising membrane integrity and function [42].
- Mechanical Stress from Ice Crystals: Extracellular ice growth can mechanically crush cells within confined channels, particularly in dense tissues and multicellular systems [2].
Cryoprotectant Protective Mechanisms
CPAs counter these damage pathways through multiple, often overlapping mechanisms:
- Colligative Action: Permeating CPAs reduce the fraction of water that freezes at any given temperature, thereby limiting solute concentration and osmotic imbalance during slow freezing [58] [42].
- Glass Formation: At sufficient concentrations, CPAs enable vitrification—a transition to an amorphous glassy state that prevents crystalline ice formation entirely during rapid cooling [52].
- Membrane Stabilization: Some CPAs, particularly disaccharides, can directly interact with phospholipid head groups, maintaining membrane integrity in dehydrated states by replacing water molecules [42].
- Ice Recrystallization Inhibition: Certain non-penetrating polymers and proteins adsorb to ice crystal surfaces, inhibiting growth and recrystallization during temperature fluctuations [58].
The following diagram illustrates the relationship between cooling rates, water transport, and the primary injury mechanisms that cryoprotectants are designed to mitigate:
Figure 1: Pathways of Freezing-Induced Cellular Damage. Slow cooling primarily causes osmotic damage through extracellular ice formation and solute concentration, while rapid cooling leads to mechanical damage through intracellular ice formation.
Cryoprotectant Classes and Properties
Cryoprotectants are broadly categorized based on their membrane permeability and molecular characteristics, which dictate their protective mechanisms and application strategies.
Penetrating Cryoprotectants
Penetrating (intracellular) CPAs are typically low molecular weight compounds that readily cross cell membranes, providing protection both inside and outside cells:
- Dimethyl Sulfoxide (DMSO): A highly effective, widely used CPA (typically at 5-10% v/v) with excellent membrane penetration but significant concentration-dependent and temperature-dependent toxicity. DMSO destabilizes proteins above 0°C but stabilizes them below 0°C [83].
- Glycerol: One of the earliest discovered CPAs, offering lower toxicity than DMSO but slower membrane permeation. Particularly effective for red blood cells, spermatozoa, and microorganisms at 5-15% concentrations [83] [84].
- Ethylene Glycol: Smaller molecular size enables faster cellular entry, beneficial for systems with permeability limitations, but exhibits higher metabolic toxicity than propylene glycol [58] [52].
- Propylene Glycol: Similar to ethylene glycol but with lower metabolic toxicity, frequently used in clinical applications including cryopreservation of oocytes and embryos [52].
Non-Penetrating Cryoprotectants
Non-penetrating (extracellular) CPAs provide protection through external mechanisms without entering cells:
- Disaccharides (Trehalose, Sucrose): Naturally occurring cryoprotectants that stabilize membranes and proteins during dehydration by forming hydrogen bonds and glassy matrices. Typically used at 0.1-0.5 M concentrations, they also serve as osmotic buffers to control cell volume during CPA addition/removal [83] [58].
- Polymers (HES, PVP, Dextran): High molecular weight compounds that increase solution viscosity, modify ice crystal growth, and provide mechanical support to cell surfaces. Often used at 2-5% w/v in combination with penetrating CPAs [58].
- Sugars (Glucose, Raffinose): Mono- and trisaccharides that function as osmotic buffers and contribute to glass formation, with specific transporters sometimes facilitating limited cellular uptake [58].
Table 1: Properties of Common Penetrating Cryoprotectants
| Cryoprotectant | Molecular Weight (g/mol) | Typical Concentration Range | Relative Toxicity | Key Applications |
|---|---|---|---|---|
| DMSO | 78.1 | 5-10% (v/v) | High | Mammalian cell lines, stem cells, therapeutic cells |
| Glycerol | 92.1 | 5-15% (v/v) | Moderate | Red blood cells, spermatozoa, microorganisms |
| Ethylene Glycol | 62.1 | 5-10% (v/v) | Moderate-High | Oocytes, embryos, sensitive cell types |
| Propylene Glycol | 76.1 | 5-10% (v/v) | Moderate | Clinical applications, oocyte preservation |
Table 2: Properties of Common Non-Penetrating Cryoprotectants
| Cryoprotectant | Molecular Class | Typical Concentration | Primary Mechanism | Key Applications |
|---|---|---|---|---|
| Trehalose | Disaccharide | 0.1-0.5 M | Glass formation, membrane stabilization | Biopharmaceuticals, RBCs, with penetrating CPAs |
| Sucrose | Disaccharide | 0.1-0.5 M | Osmotic buffer, glass former | Lyophilized formulations, CPA addition/removal |
| Hydroxyethyl Starch (HES) | Polymer | 2-5% (w/v) | Ice growth modulation, viscosity enhancement | Organ preservation, combination cocktails |
| Polyvinyl Pyrrolidone (PVP) | Polymer | 2-5% (w/v) | Surface adsorption, ice recrystallization inhibition | Sensitive cell types, research applications |
Toxicity Profiles and Mitigation Strategies
Cryoprotectant-Specific Toxicity Mechanisms
Each CPA exhibits distinct toxicity profiles that must be considered during formulation development:
- DMSO Toxicity: Induces dose-dependent membrane fluidity changes, disrupts mitochondrial function, increases reactive oxygen species, and can cause cellular differentiation. Clinical manifestations include cardiovascular instability, neurological symptoms, and allergic reactions during cell therapy infusions [83] [52]. Toxicity is markedly temperature-dependent, with significantly reduced effects at lower temperatures [83].
- Glycerol Toxicity: Generally better tolerated than DMSO but can cause osmotic stress, membrane damage at high concentrations, and renal toxicity through oxidative stress and apoptosis pathways at systemic levels [52].
- Polyol Toxicity: Ethylene glycol metabolites cause metabolic acidosis and calcium oxalate crystal formation, while propylene glycol can decrease intracellular pH at high concentrations (>2.5 M) [52].
- Formamide Toxicity: A highly corrosive amide that can denature DNA through water displacement and disrupt hydrogen bonding networks, limiting its utility despite excellent vitrification properties [52].
Formulation Strategies to Mitigate Toxicity
Successful cryoprotectant formulations implement multiple strategies to balance efficacy and toxicity:
- CPA Cocktails: Mixtures of permeating and non-permeating CPAs enable vitrification at lower individual agent concentrations, significantly reducing specific toxicities while maintaining protection [52] [85]. For example, combining DMSO with formamide, propylene glycol, and colloids creates synergistic effects that lower overall toxicity [85].
- Temperature Modulation: CPA exposure and removal procedures conducted at reduced temperatures (0-4°C) markedly decrease toxicity for many agents, particularly DMSO [83] [52].
- Stepwise Addition and Removal: Controlled, multi-step procedures for introducing and diluting CPAs prevent osmotic shock and minimize toxicity by allowing gradual cellular adaptation [83].
- Nutrient Supplementation: Cryoprotectant solutions for microorganisms and sensitive cell types may include peptone, yeast extract, and antioxidants that improve viability during preservation [84].
Table 3: Documented Toxicity Effects by Cryoprotectant Type
| Cryoprotectant | Cellular/Mechanistic Toxicity | Systemic/Clinical Toxicity | Temperature Dependence |
|---|---|---|---|
| DMSO | Membrane disruption, mitochondrial dysfunction, ROS production | Cardiovascular instability, neurological effects, allergic reactions | Strong (increased with temperature) |
| Glycerol | Osmotic stress at high concentrations, actin cytoskeleton polymerization | Renal failure with high systemic doses, oxidative stress | Moderate |
| Ethylene Glycol | Metabolic acidosis, calcium oxalate crystal formation | Gastrointestinal irritation, pulmonary edema | Weak |
| Propylene Glycol | Intracellular pH reduction at high concentrations | Few systemic effects, used in food products | Weak |
| Formamide | DNA denaturation, disruption of hydrogen bonding | Kidney injury, blood cell damage | Moderate |
Advanced Formulation Design and Optimization
CPA Cocktail Development
Modern cryopreservation protocols increasingly rely on multi-component CPA cocktails rather than single-agent formulations. These cocktails leverage complementary mechanisms of action while minimizing individual component concentrations below toxicity thresholds. The development process involves systematic evaluation of:
- Component Selection: Choosing CPAs with diverse molecular sizes, hydrogen-bonding capabilities, and toxicity profiles to create synergistic combinations [52].
- Concentration Optimization: Balancing the vitrification tendency (Cv) of each component against its specific toxicity, often requiring extensive empirical testing [52].
- Additive Supplementation: Incorporating non-protective additives that mitigate specific toxicity mechanisms or enhance membrane stability.
Recent research demonstrates that mixtures containing 70% glycerin with nutrient supplements (peptone and yeast extract) provided superior viability (88.9% survival) for Enterobacterales strains compared to glycerin alone (44.8%) after 12 months at -20°C [84]. Similarly, in tissue engineering applications, bioinks incorporating 10% glycerol demonstrated significantly improved cell viability after cryopreservation compared to CPA-free controls [82].
Protocol Optimization for Complex Biological Systems
Different biological materials require tailored formulation strategies:
- Single Cells in Suspension: Standardized protocols using 5-10% DMSO or glycerol suffice for many mammalian cell lines, with cooling rates optimized to balance solution effects and intracellular ice formation [83].
- Multicellular Systems and Tissues: Complex architectures require longer equilibration times, potential vascular delivery, and modified cocktails that address extracellular matrix preservation [52].
- Tissue-Engineered Constructs: Bioink-integrated CPAs must balance cryoprotection with maintenance of printability and mechanical properties, as demonstrated with alginate-based systems containing glycerol [82].
The following workflow outlines a systematic approach to developing and optimizing cryoprotectant formulations for new biological systems:
Figure 2: Cryoprotectant Formulation Development Workflow. A systematic approach to developing optimized cryopreservation protocols for new biological systems, incorporating iterative refinement based on functional outcomes.
Experimental Protocols and Assessment Methodologies
Standardized Viability Assessment Protocol
Robust assessment of cryopreservation outcomes requires multiple complementary assays:
- Membrane Integrity Assays: Fluorescent dye exclusion (propidium iodide) or inclusion (calcein-AM) to distinguish live/dead populations.
- Clonogenic assays: Measurement of reproductive viability through colony-forming unit efficiency post-thaw.
- Metabolic Function Assays: Resazurin reduction (alamarBlue), ATP content, or MTT conversion to assess metabolic competence.
- Functional Capacity Testing: Cell-type specific functions (differentiation potential, enzymatic activity, motility, contraction).
- Apoptosis/Necrosis Markers: Annexin V staining, caspase activation, LDH release to quantify cell death pathways.
For research requiring biochemical stability, additional assessments of protein integrity, genetic stability, and organelle function may be necessary.
Representative Experimental Protocol: Bacterial Cryopreservation
A recent systematic investigation of Enterobacterales cryopreservation provides a robust methodological template [84]:
- Inoculum Preparation: Harvest overnight cultures and prepare suspensions in phosphate-buffered saline (PBS, pH 7.2) adjusted to 0.5 McFarland units.
- Biomass Concentration: Centrifuge at 10,000 × g for 10 minutes at 20°C and resuspend pellet in cryoprotectant solutions.
- CPA Formulations Tested:
- Cryoprotectant 1: 70% glycerin + 8% glucose + nutrient supplements (peptone/yeast extract) in PBS
- Cryoprotectant 2: 10% DMSO + 70% glycerin + 8% glucose + nutrients in PBS
- Cryoprotectant 3: 10% DMSO + 8% glucose in PBS
- Cryoprotectant 4: 70% glycerin + 8% glucose in PBS
- Equilibration and Freezing: Hold suspensions at 4-6°C for 30 minutes, then transfer to -20°C storage.
- Thawing and Assessment: Rapidly thaw at 37°C for 3-5 minutes with gentle shaking. Determine viable counts using standard plate counting method after serial dilution on Nutrient Agar.
- Results: After 12 months, survival rates were 88.9% (Cryoprotectant 1), 84.9% (Cryoprotectant 2), 83.5% (Cryoprotectant 3), and 44.8% (Cryoprotectant 4), demonstrating the superiority of nutrient-supplemented glycerin formulations.
Representative Experimental Protocol: Cryobioprinting Bioink Preservation
A specialized protocol for preserving cell-laden bioinks illustrates adaptation for advanced applications [82]:
- Bioink Formulation: 4% sodium alginate dissolved in DMEM with 10% FBS and 20 mM CaCl₂ for pre-crosslinking.
- CPA Incorporation: Add DMSO (5%, 10%, 15% v/v) or glycerol (5%, 10%, 15% v/v) to pre-crosslinked bioinks.
- Rheological Assessment: Characterize viscosity and yield stress changes induced by CPA addition.
- Printability Evaluation: Conduct tube inversion and printability tests to determine optimal CPA concentrations.
- Cryopreservation and Analysis: Print constructs, preserve at -80°C for 72 hours, rapidly thaw, and assess cell viability via live/dead staining.
- Results: 10% glycerol provided optimal balance between printability maintenance and cryoprotection, significantly improving post-thaw viability compared to CPA-free controls.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for Cryoprotectant Research and Implementation
| Reagent/Category | Specific Examples | Primary Function | Application Notes |
|---|---|---|---|
| Penetrating CPAs | DMSO, Glycerol, Ethylene Glycol, Propylene Glycol | Intracellular protection, colligative freezing point depression | Use high-purity, sterile-filtered grades; concentration optimization required |
| Non-Penetrating CPAs | Trehalose, Sucrose, Raffinose | Extracellular glass formation, osmotic buffering | Consider non-reducing sugars for sensitive applications |
| Polymeric Additives | HES, PVP, Dextran, Ficoll | Ice crystal modification, viscosity enhancement | Molecular weight affects efficacy; typically used at 2-5% w/v |
| Nutrient Supplements | Peptone, Yeast Extract, FBS | Metabolic support during stress | Particularly beneficial for microbial and primary cell preservation |
| Membrane Stabilizers | Cholesterol, Phospholipids | Membrane integrity maintenance | For sensitive cell types with particular membrane compositions |
| Antioxidants | Glutathione, Ascorbic Acid | Oxidative stress reduction | Counteract ROS generation during freezing/thawing |
| Buffering Systems | PBS, HEPES, Tris | pH maintenance during CPA exchange | Critical during addition/removal steps with acidic/basic CPAs |
| Viability Assays | Propidium Iodide, Calcein-AM, AlamarBlue | Post-thaw function assessment | Multiparameter assessment recommended for complex systems |
The strategic formulation of cryoprotectant solutions represents a critical intersection of physical chemistry, cell biology, and systems engineering. Successful approaches must address both the mechanical effects of ice formation and the osmotic consequences of freeze-concentrated solutes, while simultaneously minimizing CPA-specific toxicities. The current state of the art increasingly favors multi-component cocktails that leverage complementary mechanisms of action, carefully optimized for specific biological applications through systematic empirical testing.
As cryopreservation extends to increasingly complex systems including tissue-engineered constructs, organoids, and eventually whole organs, formulation strategies must evolve to address additional challenges of mass transport, heterogeneous cell populations, and functional preservation. The integration of computational modeling, high-throughput screening, and molecular design approaches holds promise for developing next-generation cryoprotectants with enhanced efficacy and reduced toxicity. Through continued refinement of these formulation strategies, researchers can expand the boundaries of biological preservation, enabling advances in regenerative medicine, bioproduction, and fundamental biological research.
Cryoprotective agents (CPAs) are essential for successful cell and organ cryopreservation, yet their introduction and removal create significant osmotic stress that can compromise cellular integrity and function. During CPA introduction, water rapidly exits cells causing initial shrinkage, followed by CPA influx and subsequent water return that can lead to excessive swelling and damage. The reverse process occurs during CPA removal, with water influx causing swelling followed by CPA efflux and consequent shrinkage. Managing these volume excursions is critical for maintaining membrane integrity, cellular function, and post-preservation viability. This technical guide examines the fundamental principles and advanced methodologies for controlling osmotic stress within the broader context of mechanical and osmotic effects of freezing on cellular systems, providing researchers with evidence-based strategies for optimizing cryopreservation protocols across biological scales from individual cells to whole organs.
Fundamental Principles of Osmotic Stress
Physicochemical Basis of Osmotic Injury
Cellular response to osmotic stress is governed by the Kedem-Katchalsky equations derived from irreversible thermodynamics, which describe the coupled transport of water and solutes across semi-permeable membranes [86]. The volumetric flow rate (Jv) and CPA flow rate (Jcpa) are mathematically described as:
Jv = S·Lp[(Pf-Pt)-RgT(Cis,f-Cis,t+σ{Ccpa,f-Ccpa,t})]
Jcpa = S[ωRgT(Ccpa,f-Ccpa,t)]+Jv(1-σ)(Ccpa,f+Ccpa,t)/2
Where S represents membrane surface area (m²), Lp is hydraulic conductivity [m³/(N·s)], P is hydraulic pressure (N/m²), Rg is the universal gas constant [J/(mol·K)], T is temperature (K), C is concentration (mol/m³), ω is CPA permeability [mol/(N·s)], and σ is the reflection coefficient [86]. These equations highlight that osmotic damage occurs through two primary mechanisms: (1) mechanical damage from excessive volume excursions beyond tolerable limits, and (2) chemical toxicity from CPA exposure at specific concentrations and durations.
Cellular Tolerance Limits
Cellular membranes can typically withstand volume changes of 50-60% without irreversible damage, though this varies significantly by cell type. The table below summarizes critical osmotic parameters for common experimental cell models:
Table 1: Key Osmotic Parameters for Common Cell Types
| Cell Type | Hydraulic Conductivity (Lp) ×10⁻¹⁴ m³/(N·s) | CPA Permeability (ω) ×10⁻¹³ mol/(N·s) | Reflection Coefficient (σ) | Maximum Tolerable Volume Change |
|---|---|---|---|---|
| RAW264.7 Macrophages | 1.5 [8] | 7.0 [8] | 0.10 [8] | ~55% [8] |
| Rat Kidney Cells | 1.5 [86] | 7.0 [86] | 0.10 [86] | ~60% [86] |
| LGG Probiotics | Not specified | Not specified | Not specified | ~50% [87] |
| Huh-7 Hepatocytes | Not specified | Not specified | Not specified | ~50% [88] |
Mathematical Modeling of CPA Transport
Krogh Cylinder Model for Organ Systems
The Krogh cylinder model provides a theoretical framework for analyzing CPA transport in vascularized tissues by representing the organ as repeating functional units, each containing a central capillary surrounded by tissue cells [86]. This approach is particularly valuable for modeling whole organ perfusion, where mass transfer primarily occurs at the capillary level due to the high surface-area-to-volume ratio. The model enables researchers to predict CPA diffusion and concentration profiles throughout tissues, accounting for variables such as flow rates, temperature, and capillary density. When combined with toxicity cost functions, this integrated approach allows for protocol optimization that minimizes both osmotic and chemical damage [86].
Toxicity Cost Function Model
The toxicity cost function model quantifies cumulative CPA toxicity as a function of exposure concentration, time, and temperature through a power law relationship [86]:
k = β·Cαcpa,t
dN/dt = -k·N
Jtox = ∫β·Cαcpa,tdt
N/N₀ = exp(-Jtox)
Where k represents the toxicity rate (1/min), α and β are constants dependent on CPA formulation and biological system, Ccpa,t is tissue CPA concentration, N is viability after exposure time tf, N₀ is initial viability, and Jtox is the toxicity cost function quantifying cumulative toxicity [86]. For VMP (a common CPA cocktail containing DMSO, formamide, ethylene glycol, and ice blockers) in rat kidneys, parameters were measured as α = 3.12 and β = 9.39 × 10⁻⁶ [86]. This model enables researchers to optimize protocols by balancing sufficient CPA concentration for vitrification against time-dependent toxic effects.
Experimental Protocols for Osmatic Stress Management
Controlled-rate perfusion protocols for CPA introduction and removal are essential for managing osmotic stress. The following optimized protocol for rat kidney perfusion with VMP demonstrates this approach [86]:
Table 2: Optimized VMP Perfusion Protocol for Rat Kidneys
| Step | Solution | Duration (min) | Temperature (°C) | Flow Rate (mL/min) | Objective |
|---|---|---|---|---|---|
| Baseline | Standard Perfusate | 10 | 4 | 15 | Establish baseline function |
| CPA Introduction 1 | 1.5M VMP | 5 | 4 | 12 | Moderate initial osmolarity increase |
| CPA Introduction 2 | 3.0M VMP | 7 | 4 | 10 | Gradual concentration increase |
| CPA Introduction 3 | 4.5M VMP | 6 | 4 | 8 | Approach target concentration |
| CPA Introduction 4 | 6.0M VMP | 4 | 4 | 6 | Final concentration step |
| Equilibration | 8.4M VMP | 3 | 4 | 5 | Achieve uniform distribution |
| Total CPA Exposure | 25 minutes | Minimized toxicity |
This optimized protocol reduced total exposure time by 18.5% compared to traditional empirical protocols while maintaining comparable physical and biological outcomes [86]. The stepwise approach allows cells to gradually adjust to changing osmotic conditions, minimizing volume excursions beyond critical thresholds.
Advanced CPA Formulations for Reduced Stress
Emerging CPA technologies focus on membrane-targeted approaches that provide cryoprotection with reduced osmotic stress. Cholesterol-functionalized DNA frameworks (Chol24-DF) represent one innovative strategy, offering several advantages [8]:
- Targeted membrane interaction through cholesterol anchoring reduces required intracellular CPA concentrations
- Enhanced ice recrystallization inhibition at membrane interfaces where ice formation is most damaging
- Autonomous biodegradation upon thawing eliminates post-preservation toxicity concerns
- Modular design enables incorporation of additional functional moieties for specific cell types
In macrophage cell lines (RAW264.7), Chol24-DF demonstrated clear advantages over conventional DMSO, protecting frozen cells through enhanced membrane targeting while maintaining viability, morphology, apoptosis levels, metabolism (ATP levels), and innate immune function (nitric oxide production) after cryopreservation [8].
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Research Reagent Solutions for Osmatic Stress Management
| Reagent/Material | Function | Application Notes | Key References |
|---|---|---|---|
| VMP Cocktail | Multi-component CPA | Contains DMSO, formamide, ethylene glycol, X-1000, Z-1000; optimized for kidney vitrification | [86] |
| Chol24-DF | Membrane-targeted DNA framework | Hexagonal wireframe structure with cholesterol anchors; biodegradable after thawing | [8] |
| Synth-a-Freeze Medium | Chemically defined cryopreservation medium | Protein-free, contains 10% DMSO; suitable for stem and primary cells | [7] |
| Recovery Cell Culture Freezing Medium | Complete cryopreservation medium | Optimized FBS to BSA ratio for improved viability and recovery | [7] |
| Trehalose-Skim Milk Protectant | Cryoprotectant for probiotics | 5-10% concentration; forms microcapsule structures around bacterial cells | [87] |
| Controlled-Rate Freezer | Temperature management apparatus | Maintains cooling rate of approximately 1°C/minute to minimize ice crystal formation | [7] |
| Krogh Cylinder Model | Computational transport model | Predicts CPA distribution and osmotic stress in vascularized tissues | [86] |
Technical Protocols for Specific Applications
Cell Freezing Protocol for Mammalian Cells
The standard protocol for cryopreserving mammalian cells while managing osmotic stress involves these critical steps [7]:
- Harvest log-phase cells at at least 90% viability using gentle detachment methods for adherent cells
- Centrifuge at 100-400 × g for 5-10 minutes and resuspend in cold freezing medium
- Use controlled freezing apparatus to reduce temperature at approximately 1°C per minute
- Store in liquid nitrogen vapor phase below -135°C to preserve viability
For osmotic stress management specifically, the freezing medium composition is critical. Traditional formulations include complete medium with 10% DMSO or glycerol, or serum-free alternatives containing 7.5% DMSO with 10% cell culture-grade BSA [7]. The stepwise addition of CPA-containing media to cell suspensions, rather than direct exposure to final concentrations, significantly reduces osmotic shock.
Freeze-Drying Protocol for Single-Cell Imaging
For ToF-SIMS single-cell imaging, this optimized freeze-drying protocol maintains cellular morphology while managing osmotic stress during water removal [88]:
- Wash cells in PBS for 2-3 seconds, repeated twice
- Transfer to 0.15M ammonium formate solution for approximately 30 seconds
- Rinse sequentially in three separate tubes to ensure complete salt removal
- Rapid freezing in isopentane coolant pre-cooled with liquid nitrogen
- Lyophilize at -55°C and 10⁻³ mbar pressure for 12 hours
- Gradually warm to ambient temperature to evaporate residual isopentane
This protocol specifically addresses osmotic stress during the freezing and dehydration phases by controlling ice crystal formation through rapid freezing and using ammonium formate to stabilize membranes during solute concentration changes [88].
Effective management of osmotic stress during CPA introduction and removal requires integrated strategies combining mathematical modeling, controlled physical parameters, and advanced CPA formulations. The Krogh cylinder model combined with toxicity cost functions provides a powerful framework for optimizing perfusion protocols, particularly for complex tissues and organs. Stepwise introduction and removal of CPAs with careful attention to concentration gradients, exposure times, and temperature parameters significantly reduces osmotic injury. Emerging technologies such as membrane-targeted DNA frameworks offer promising alternatives to traditional CPAs by providing effective cryoprotection with reduced osmotic stress and inherent biodegradability. As cryopreservation applications expand to include increasingly complex biological systems from organoids to whole organs, precise management of osmotic stress will remain a critical factor determining preservation success and post-thaw functionality.
Cold Acclimation and Membrane Lipid Composition Modification
Within the broader context of research on the mechanical and osmotic effects of freezing on cells, the process of cold acclimation represents a critical biological adaptation. When cells encounter freezing temperatures, the formation of extracellular ice creates profound mechanical stresses and osmotic imbalances. The growth of ice crystals can physically damage cellular structures, while the resultant dehydration concentrates intracellular solutes to toxic levels [42]. A primary site of this damage is the plasma membrane, whose integrity is compromised by the close apposition of membranes and lipid rigidification [89] [42]. Cold acclimation encompasses the morphological, physiological, biochemical, and molecular changes that organisms undergo to prepare for and survive these low-temperature challenges [90]. This review examines how modification of membrane lipid composition serves as a fundamental mechanism conferring freezing tolerance within this mechanical and osmotic framework.
Cold Sensing and Initial Signaling Events
The cold acclimation process initiates with the perception of temperature decrease, triggering a signaling cascade that ultimately coordinates changes in gene expression and membrane composition.
Membrane Rigidification as the Primary Signal
The earliest event in cold sensing is a change in membrane physical state. A drop in temperature rigidifies the phospholipid bilayers of cellular membranes, reducing fluidity and compromising membrane function [91]. This rigidification provides the physical stimulus for downstream signaling pathways.
Calcium-Mediated Signaling Cascade
Membrane rigidification activates plasma membrane calcium channels, leading to an influx of Ca²⁺ into the cytoplasm [91]. The frequency, duration, and amplitude of these calcium signals encode information about the severity of cold stress. These calcium signatures are detected by calcium-binding proteins including calmodulin (CaM), CaM-like proteins (CML), calcium-dependent protein kinases (CDPKs), and calcineurin B-like proteins (CBLs) [91]. This signaling network ultimately regulates the expression of cold-responsive genes through transcription factors such as Inducer of CBF Expression 1 (ICE1) and C-repeat Binding Factors (CBFs) [91].
The following diagram illustrates this cold sensing and signaling pathway:
Cold Sensing and Signaling Pathway
Membrane Lipid Modifications During Cold Acclimation
Homeoviscous Adaptation
A central lipid modification in response to cold is homeoviscous adaptation, a process whereby organisms adjust membrane lipid composition to maintain fluidity and functionality at lower temperatures [89]. This process involves changing the saturation degree of phospholipid acyl chains, altering the proportion of different phospholipid classes, and modifying sterol content. These changes counter the rigidifying effect of cold temperatures, ensuring proper membrane protein function and transport processes.
Quantitative Changes in Lipid Composition
Cold acclimation induces specific, quantifiable alterations in membrane lipid profiles. The following table summarizes key lipid modifications documented in experimental systems:
Table 1: Membrane Lipid Modifications in Response to Cold Acclimation
| Lipid Parameter | Change During Cold Acclimation | Functional Significance | Experimental System |
|---|---|---|---|
| Phospholipid Unsaturation | Increased proportion of unsaturated fatty acids | Enhances membrane fluidity at low temperatures; prevents phase transitions | Insects, Plants [89] |
| Phosphatidylcholine (PC) Species | Increased polyunsaturated species (e.g., PC 18:3/16:3) | Promotes non-vesicular transport to ER; faster retrograde trafficking | Mammalian Cells [92] |
| Sphingomyelin (SM) Content | Altered distribution and metabolism | Affects lipid raft stability and signaling microdomains | Mammalian Cells [92] |
| Antifreeze Glycolipids | Production of specific glycolipid species | Inhibits ice crystal growth and recrystallization | Insects [89] |
| Cuticular Lipids | Increased accumulation | Provides extra insulation and barrier protection | Insects [89] |
These lipid modifications collectively serve to preserve membrane integrity and function under freezing conditions. The increased proportion of unsaturated phospholipids maintains membrane fluidity, while specialized lipids like antifreeze glycolipids provide mechanical protection against ice crystal damage [89]. The specific changes in PC species composition directly impact intracellular lipid trafficking, with polyunsaturated species showing faster retrograde transport from the plasma membrane to the endoplasmic reticulum [92].
Experimental Approaches for Studying Cold-Induced Lipid Changes
Quantitative Imaging of Lipid Transport
Recent advances enable precise quantification of lipid dynamics in living cells during cold stress. The following workflow illustrates a comprehensive approach for imaging lipid transport and metabolism:
Lipid Transport Imaging Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Studying Cold Acclimation and Membrane Lipids
| Reagent/Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| Cryoprotective Agents | DMSO, Glycerol, Synth-a-Freeze Medium | Reduce freezing point; slow cooling rate; prevent ice crystal formation | Use at 5-10% concentration; DMSO facilitates organic molecule entry [7] |
| Bifunctional Lipid Probes | Diazirine- and alkyne-modified PC, PE, PA, SM | Enable tracking of lipid localization and metabolism via click chemistry | Minimal modifications preserve native lipid behavior; resemble palmitoleic acid [92] |
| Membrane Staining Reagents | Organelle markers (ER, Golgi, endosomes, mitochondria) | Assign lipid signals to specific organelles in imaging studies | Use Ilastik software for segmentation and probability mapping [92] |
| Cell Freezing Media | Recovery Cell Culture Freezing Medium, Serum-free formulations | Preserve cell viability during cryopreservation | Contain cryoprotectant + protein source; controlled-rate freezing recommended [7] |
| Lipid Analysis Tools | Ultra-high-resolution FT Mass Spectrometry, Shotgun Lipidomics | Quantify lipid species and metabolic conversions | Distinguish bifunctional lipids via mass difference of diazirine group [92] |
Freeze-Fracture Replica Methods for Membrane Analysis
The freeze-fracture replica technique with SDS digestion provides high-resolution analysis of membrane lipid and protein distribution. This method involves rapid freezing of samples, fracturing to reveal membrane interiors, and creating carbon/platinum replicas [93] [94]. Quantitative retention of membrane lipids in these replicas enables immunogold labeling to determine localization of specific lipid species, with carbon-first evaporation providing superior labeling efficiency for ganglioside GM1 and phosphatidylcholine [94].
Implications for Cryopreservation and Drug Development
Understanding natural cold acclimation mechanisms provides valuable insights for developing improved cryopreservation protocols and pharmaceutical approaches. The principles of homeoviscous adaptation directly inform the composition of cryopreservation media, with specific lipid mixtures potentially enhancing cell survival during freezing and thawing [7]. For drug development, membrane lipid modifications represent potential therapeutic targets for conditions involving cold exposure or ischemic injury, where similar mechanical and osmotic stresses occur.
The recognition that non-vesicular lipid transport dominates the organelle distribution of lipids [92] suggests new strategies for manipulating cellular responses to cold stress. Furthermore, the production of antifreeze proteins and glycolipids during cold acclimation [91] offers biomimetic inspiration for cryoprotectant development, with potential applications in organ preservation and regenerative medicine.
Cold acclimation through membrane lipid composition modification represents a sophisticated biological response to the mechanical and osmotic challenges of freezing. By activating specific signaling pathways that ultimately reprogram lipid metabolism, organisms can restructure their membranes to maintain functionality under temperature extremes. The experimental approaches detailed here provide powerful tools for quantifying these changes, while the growing understanding of lipid trafficking mechanisms offers new avenues for biomedical application. Within the broader context of freezing effects on cells, these adaptive mechanisms highlight the crucial role of membrane homeostasis in surviving cold stress.
Controlling Ice Nucleation Temperature and Crystal Size Distribution
The controlled freezing of biological materials is a cornerstone of modern bioscience, drug development, and cryopreservation. At the heart of this process lies the precise management of ice nucleation—the initial formation of ice crystals—and the subsequent growth that determines crystal size distribution. Uncontrolled ice formation is a primary source of cell damage during freezing, arising through two interconnected mechanisms: direct mechanical injury from ice crystals piercing cellular structures, and osmotic stress induced by solute concentration in unfrozen fractions [73] [95]. This technical guide provides an in-depth examination of the factors governing ice nucleation and crystal growth, with a specific focus on mitigating these damaging effects on cellular systems. By integrating fundamental theory, experimental data, and practical methodologies, this review equips researchers with the tools to optimize freezing protocols for preserving cell viability and function.
Theoretical Foundations
Classical Nucleation Theory and Ice Formation
Ice nucleation is an activated process that requires surmounting a free energy barrier, described by Classical Nucleation Theory (CNT). This barrier exists because the formation of a new, stable ice phase necessitates the creation of an interface between the solid ice and liquid water, which carries an associated interfacial free energy [96].
The nucleation rate ((J)), representing the frequency of nucleation events per unit volume, is given by: [ J = A N0 \exp\left(-\frac{\Delta G^*}{kT}\right) ] where (A) is a kinetic constant, (N0) is the concentration of potential nucleation sites, (\Delta G^) is the activation barrier (nucleation work), (k) is the Boltzmann constant, and (T) is the absolute temperature [96]. The activation barrier (\Delta G^) is the sum of a bulk free energy term (which favors nucleation and is proportional to the volume of the nucleus) and an interfacial free energy term (which opposes nucleation and is proportional to the surface area of the nucleus) [96]. The size at which an ice nucleus becomes stable and can continue to grow is known as the critical nucleus size ((r^*)) [96].
In practice, homogeneous nucleation (formation of ice in pure water without external influences) rarely occurs until temperatures reach -40°C or below due to the exceptionally high energy barrier [97]. Most freezing processes involve heterogeneous nucleation, where the presence of foreign surfaces or particles catalyzes ice formation at significantly higher temperatures by reducing the activation barrier [96] [97].
Mechanisms of Freezing Injury to Cells
The formation of ice extracellularly initiates a cascade of potentially lethal events for cells, primarily through two interconnected mechanisms:
- Osmotic Stress: As extracellular ice forms, dissolved solutes are excluded from the crystal lattice, leading to a dramatic increase in the solute concentration of the remaining unfrozen fraction. This creates a steep osmotic gradient across the cell membrane, causing water to efflux from the cell interior. This dehydration can concentrate intracellular solutes to toxic levels, compress the plasma membrane, and potentially lead to lipoprotein subunit dissociation in membrane structures [73].
- Mechanical Stress: Intracellular ice formation, which typically occurs at rapid cooling rates, can cause direct mechanical damage to organelles and membrane systems. Even when ice is confined to extracellular spaces, the growing crystals can mechanically compress and physically rupture cells [73] [95].
Research on winter rye leaves demonstrates that exposure to hypertonic sorbitol solutions can cause injury equivalent to freezing to subzero temperatures that produce the same osmotic stress, underscoring the critical role of osmotic imbalance in freezing injury [95].
Factors Influencing Ice Nucleation and Crystal Growth
Key Experimental Parameters
Recent investigations using Differential Scanning Calorimetry (DSC) have systematically quantified how various experimental parameters affect the ice nucleation temperature distribution [97]. The median nucleation temperature ((T{50})) and the distribution width ((T{10}-T_{90}), the temperature difference between the 10th and 90th percentiles of the survival curve) are critical metrics for characterizing nucleation behavior.
Table 1: Effect of Experimental Parameters on Ice Nucleation Temperature Distribution
| Parameter | Effect on Median Nucleation Temperature ((T_{50})) | Effect on Distribution Width ((T{10}-T{90})) | Mechanistic Insight |
|---|---|---|---|
| Thawing Temperature ((T_{th})) | Negligible direct effect [97] | Increases significantly with higher (T_{th}) (e.g., from 20°C to 60°C) [97] | Enhanced molecular diffusion at higher temperatures alters the configuration/position of nucleating agents. |
| Residence Time Above 0°C ((\tau)) | Negligible direct effect [97] | Increases with longer (\tau) (from ~2°C at 5 min to ~8°C at 60 min) [97] | Longer times allow for increased diffusion of nucleators, increasing stochasticity. |
| Cooling Rate | Generally decreases with faster cooling [97] | Can be correlated with (\tau) in experimental design [97] | Less time for nucleation events at specific temperatures; affects crystal size. |
| Sample Volume | Increases with larger volumes [97] | -- | Larger volumes contain more potential nucleation sites. |
| Nucleating Agents | Increases significantly (reduces supercooling) [96] [97] | Depends on agent concentration and properties [97] | Provides templates for ice formation, lowering activation energy. |
The Role of Diffusion Kinetics
A pivotal finding in recent nucleation studies is the profound influence of diffusion kinetics on nucleation temperature distributions. Both the thawing temperature and the residence time above 0°C between freeze-thaw cycles dictate the level of thermal agitation and diffusion of potential nucleators within the sample [97].
Conditions that enhance diffusion—such as higher thawing temperatures or longer residence times—result in broader, more scattered nucleation temperature distributions. Conversely, conditions that limit diffusion—such as lower thawing temperatures or shorter residence times—produce narrower, more regular distributions [97]. This relationship is formalized in the concept of residence time ((\tau)), which incorporates both the holding time at the thawing temperature ((th)) and the time spent heating to that temperature: (\tau = th + \frac{2T_{th}}{R}) [97]. This parameter provides a unified metric for predicting and controlling nucleation stochasticity.
Methodologies for Controlled Freezing in Biological Systems
Standard Cell Cryopreservation Protocol
The following protocol outlines a generalized method for cryopreserving cultured mammalian cells, incorporating steps to manage nucleation and minimize ice crystal damage [7].
- Pre-freezing Preparation: Cells should be in log-phase growth and characterized for high viability (>90%) prior to freezing. Use cells at as low a passage number as possible [7].
- Harvesting: For adherent cells, gently detach using a suitable dissociation reagent like trypsin. Resuspend the cell pellet in a complete growth medium [7].
- Cryoprotective Agent (CPA) Addition: Centrifuge the cell suspension (100–400 × g for 5–10 minutes), aspirate the supernatant, and resuspend the cell pellet in pre-chilled freezing medium. The standard freezing medium consists of a complete growth medium supplemented with 10% DMSO or a specialized commercial cryopreservation medium [7].
- Aliquoting: Dispense the cell suspension into sterile cryovials, mixing gently but frequently to maintain a homogeneous cell suspension [7].
- Controlled-Rate Freezing: Freeze the cells slowly at a rate of approximately –1°C per minute. This can be achieved using a controlled-rate cryo-freezer or an isopropanol freezing chamber placed at –80°C overnight [7].
- Long-Term Storage: Transfer the frozen cryovials to a liquid nitrogen storage tank, ideally in the vapor phase (below –135°C) to minimize explosion risks associated with liquid-phase storage [7].
Experimental Techniques for Nucleation Studies
For fundamental research into nucleation behavior, Differential Scanning Calorimetry (DSC) provides a powerful tool for precise thermal analysis.
- DSC Experimental Workflow: The general method involves performing numerous (e.g., 40-130) freeze-thaw cycles on a water sample within a DSC pan. Each cycle involves cooling from a thawing final temperature ((T{th})) to a freezing final temperature ((Tf)) and back, at specified cooling/heating rates ((R)). The sample is held at (T{th}) for a defined time ((th)) to ensure complete melting [97].
- Data Analysis: The nucleation temperature ((Tn)) for each cycle is identified from the exothermic freezing peak. The cumulative data from all cycles are plotted as a survival curve, from which the median nucleation temperature ((T{50})) and distribution width ((T{10}-T{90})) are calculated [97].
The following diagram illustrates the logical relationship between experimental parameters, diffusion kinetics, and the resulting ice nucleation characteristics, as revealed by DSC studies.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagents and Materials for Controlled Freezing Experiments
| Item | Function/Role | Example/Notes |
|---|---|---|
| Cryoprotective Agents (CPAs) | Reduce freezing point, slow cooling rate, inhibit intracellular ice crystal formation, and mitigate osmotic shock [7]. | Dimethyl Sulfoxide (DMSO) at 7.5-10%, Glycerol at 10% [7]. |
| Specialized Freezing Media | Provides a optimized, ready-to-use formulation to maximize post-thaw cell viability and recovery. | e.g., Gibco Recovery Cell Culture Freezing Medium (with serum) or Gibco Synth-a-Freeze (serum-free, chemically defined) [7]. |
| Controlled-Rate Freezing Apparatus | Enables precise, reproducible linear cooling at ~1°C/min, critical for successful cryopreservation [7]. | Controlled-rate cryo-freezers or simple isopropanol chambers (e.g., "Mr. Frosty") [7]. |
| Cryogenic Storage Vials | Secure, leak-proof containment for samples during freezing and long-term storage. | Sterile, internally-threaded cryovials suitable for liquid nitrogen temperatures [7]. |
| Differential Scanning Calorimetry (DSC) | Quantifies thermal transitions (e.g., nucleation temperature, melting point) and measures nucleation statistics [97]. | Used for fundamental studies on nucleation behavior and CPA efficacy [97]. |
Discussion and Research Implications
Integrating Osmotic and Mechanical Stress Perspectives
The parallel between injury from hypertonic solutions and freezing, as demonstrated in winter rye plasma membrane studies [95], provides a powerful framework for understanding freezing injury. The plasma membrane is a primary site of freezing damage, with both freezing and hypertonic stress causing similar increases in membrane permeability and alterations to ATPase activity and polypeptide profiles [95]. This confirms that osmotic stress is a dominant mechanism of freezing injury.
Controlling ice nucleation temperature and crystal size distribution directly addresses both osmotic and mechanical stress. Promoting a single, relatively high nucleation temperature (to minimize supercooling) results in a more controlled progression of the freezing front, reducing mechanical stresses from rapid ice growth. Simultaneously, it allows for a more predictable solute concentration profile in the unfrozen fraction, enabling cells to adapt their volume regulation more effectively [73] [95] [97].
Strategic Control of Nucleation for Cell Viability
The insights into diffusion kinetics [97] have direct practical implications. For reproducible results in freezing experiments, precisely controlling not only the cooling rate but also the thermal history of the sample before freezing—namely the thawing temperature and residence time—is critical. To maximize consistency in nucleation temperature, samples should be equilibrated at a consistently low (T_{th}) with a short (\tau) between experimental runs.
The use of cryoprotective agents like DMSO and glycerol remains fundamental. These compounds do not solely function by lowering the freezing point; they also reduce the amount of ice formed at any given temperature, thereby directly lessening the osmotic stress on cells [73] [7]. Furthermore, they can modify ice crystal morphology and growth kinetics, contributing to the overall reduction of mechanical damage.
Future Research Directions
Significant challenges and opportunities remain in this field. A major outstanding issue is the lack of a complete molecular-level picture of ice nucleation at interfaces [96]. Furthermore, the limitations of Classical Nucleation Theory in fully predicting observed nucleation behaviors necessitate the development of more comprehensive models [96]. Future research should focus on:
- Discovery of Novel Ice-Nucleating Agents and Cryoprotectants: Identifying compounds that can reliably trigger nucleation at specific, elevated temperatures would revolutionize cryopreservation protocols.
- Elucidating the Direct Molecular Interaction between cryoprotectants, water molecules, and biological membranes to design more effective protective strategies.
- Leveraging the link between diffusion kinetics and nucleation stochasticity to develop protocols that either exploit this variability (e.g., in selective freezing applications) or suppress it entirely for maximum reproducibility.
Controlling ice nucleation temperature and crystal size distribution is not merely a technical goal but a fundamental requirement for advancing research into the mechanical and osmotic effects of freezing on cells. The ability to dictate when and how ice forms enables researchers to decouple and study the individual stress pathways that lead to cell injury. By integrating an understanding of classical nucleation theory, the impact of diffusion kinetics on nucleation stochasticity, and the well-established protocols for cell cryopreservation, scientists can design more robust and reproducible freezing experiments. This integrated approach is essential for progress in diverse fields, from fundamental cell biology and the development of novel biopharmaceuticals to the long-term preservation of tissues and organs.
Thawing Process Optimization to Prevent Devitrification and Recrystallization
The process of thawing cryopreserved cells is not merely a reversal of freezing but a critical phase where devitrification and ice recrystallization can inflict severe mechanical and osmotic damage, compromising cell viability and function. Within the broader context of research on the mechanical and osmotic effects of freezing on cells, optimizing the thawing process is paramount for ensuring the reliability of biological research and the efficacy of cell-based therapies. While cryopreservation aims to suspend biological time, the thawing process presents unique challenges; ice recrystallization—the growth of larger ice crystals at the expense of smaller ones during warming—can cause significant mechanical damage to cell membranes and organelles [98] [79]. Simultaneously, cells experience profound osmotic stress as the extracellular environment changes rapidly during ice melt [99]. For researchers and drug development professionals, controlling these phenomena is not an academic exercise but a practical necessity to achieve consistent, high-quality results in experiments and therapeutics. This guide provides a detailed examination of the underlying mechanisms and presents optimized, actionable protocols to mitigate these risks.
Fundamental Damage Mechanisms During Thawing
Ice Recrystallization and Devitrification
During thawing, two interrelated physical processes pose the greatest threat to cell viability: ice recrystallization and devitrification.
Ice Recrystallization: This process occurs when small, unstable ice crystals melt and re-freeze onto larger, more stable ice crystals during temperature fluctuations, a common occurrence during slow or non-uniform warming [79]. This recrystallization leads to the formation of larger, more damaging ice crystals that can puncture cell membranes and disrupt intracellular structures [98]. The damage is particularly acute during Transient Warming Events (TWEs), where even brief temperature excursions can trigger significant ice crystal growth [98].
Devitrification: During rapid cooling, the intracellular and extracellular solutions can form an amorphous, glassy state (vitrification) instead of crystalline ice. Upon warming, if the temperature increases too slowly through a critical range, this glassy state can de-vitrify and form ice crystals de novo [100]. This devitrification process generates ice crystals in locations and morphologies that are particularly damaging to delicate cellular structures.
The following table summarizes the key characteristics of these damaging processes:
Table 1: Characteristics of Ice-Related Damage Mechanisms During Thawing
| Damage Mechanism | Trigger Condition | Primary Effect on Cells | Resultant Cellular Injury |
|---|---|---|---|
| Ice Recrystallization | Slow or fluctuating warming rates; Transient Warming Events (TWEs) [98] [79] | Growth of large, sharp extracellular and intracellular ice crystals | Mechanical shearing of plasma membranes and organelle membranes [98] |
| Devitrification | Slow warming through the "glass transition" temperature range [100] | Spontaneous crystallization from a vitrified (glassy) state | Intracellular ice formation causing irreversible structural damage [100] |
| Osmotic Shock | Rapid influx of water into dehydrated cells as extracellular ice melts [99] [101] | Swelling and potential rupture of the cell membrane | Loss of cytoplasmic components, membrane integrity failure [101] |
Osmotic and Ionic Stressors
Beyond mechanical ice damage, cells face significant osmotic and ionic challenges during thawing. As ice melts, the extracellular environment transitions from a hypertonic to a hypotonic state. This creates a strong osmotic gradient that drives water rapidly into the partially dehydrated cells [99]. If this influx is too rapid, the cells may swell beyond their volumetric tolerance and lyse—a phenomenon known as osmotic shock [101].
Recent research has uncovered an additional damaging mechanism: freezing-driven ionic charge imbalance. As ice forms during freezing, chloride ions are more readily incorporated into the ice lattice than sodium ions, creating a charge imbalance in the residual liquid phase [99]. This imbalance can generate a transient electric pulse across cell membranes during thawing, increasing membrane permeability and potentially leading to pore formation and exacerbated osmotic injury [99]. Molecular dynamics simulations have confirmed that this ionic imbalance can cause significant disruption to lipid membranes, facilitating water penetration and pore formation [99].
Diagram 1: Thawing Damage Pathways
Experimental Protocols for Thawing Optimization
Standardized Rapid Thawing Protocol
A robust and widely applicable thawing protocol is essential for minimizing devitrification and recrystallization. The following step-by-step methodology is optimized for maximum cell recovery:
Preparation: Pre-warm complete growth medium to 37°C or the appropriate temperature for the specific cell type. Prepare a centrifuge set to 1500 rpm (approximately 100–400 × g) for 5–10 minutes [7] [101].
Rapid Thawing: Remove the cryovial from liquid nitrogen storage and immediately place it in a 37°C water bath. Gently agitate the vial to ensure uniform warming. The thawing process should be rapid, taking approximately 60–90 seconds. Critical step: Remove the vial from the water bath while a small ice crystal remains in the solution [101]. This ensures the cells are not exposed to elevated temperatures for prolonged periods and prevents the toxic effects of DMSO at room temperature [101].
Dilution and Cryoprotectant Removal: Immediately after thawing, wipe the cryovial with 70% ethanol to prevent contamination. Gently transfer the cell suspension to a sterile tube containing 10 mL of pre-warmed medium. This rapid dilution is crucial to reduce the concentration of cytotoxic cryoprotectants like DMSO [101].
Gentle Centrifugation: Centrifuge the cell suspension at 1500 rpm for 5 minutes to pellet the cells and remove the cryoprotectant-containing supernatant. Avoid high-speed centrifugation, which can damage already stressed cells [101].
Resuspension and Seeding: Resuspend the cell pellet in fresh, pre-warmed complete growth medium. Count cells using a hemocytometer or automated cell counter with Trypan Blue exclusion to assess viability [7]. Seed the cells at the recommended density for the specific cell line, ensuring optimal cell-to-cell contact for recovery and proliferation [101].
Protocol for Assessing Thawing Efficacy
To systematically evaluate and optimize thawing protocols for specific cell types, the following experimental methodology can be employed:
Variable Application: Thaw identical cell aliquots using different warming rates (e.g., 37°C water bath vs. room temperature vs. 4°C) and different post-thaw handling conditions (e.g., immediate dilution vs. delayed dilution) [101].
Viability Assessment: Immediately post-thaw, determine cell viability using Trypan Blue exclusion or fluorescent viability stains like SYTO 13/GelRed [65]. This provides an initial assessment of membrane integrity.
Functional Assays: Culture the thawed cells and assess functional outcomes. For adherent cells, evaluate attachment efficiency at 24 hours post-thaw. For all cell types, measure proliferation rates over several days and assess phenotype-specific functions (e.g., stem cell differentiation capacity, immune cell activation) [98]. These delayed functional assays are critical, as cells may appear viable immediately post-thaw but undergo delayed onset cell death (DOCD) due to accumulated stress [98].
Ice Crystal Analysis: For mechanistic studies, use low-field nuclear magnetic resonance (LF-NMR) to analyze water status and distribution in thawed samples, or microscopy techniques to visualize ice crystal morphology and tissue damage indirectly [79].
Table 2: Key Reagents and Equipment for Thawing Optimization Studies
| Item Category | Specific Examples | Function & Importance |
|---|---|---|
| Cryoprotectant Agents | Dimethyl sulfoxide (DMSO), Glycerol, Synth-a-Freeze Cryopreservation Medium [7] | Permeate cells to suppress ice formation and reduce osmotic shock; require careful dilution during thawing [101] |
| Viability Assessment Tools | Trypan Blue, Automated Cell Counters (e.g., Countess), Fluorescent stains (SYTO 13/GelRed) [7] [65] | Provide quantitative assessment of post-thaw membrane integrity and cell survival |
| Specialized Equipment | Controlled-rate freezer, 37°C water bath, Low-speed centrifuge, Liquid nitrogen storage system [7] | Ensure consistent, reproducible thawing conditions and cold chain maintenance |
| Ice Recrystallization Inhibitors | Antifreeze proteins, Polyvinyl alcohol (PVA), Glycoproteins [99] [98] | Nature-inspired molecules that inhibit the growth of ice crystals during transient warming events [98] |
| Cell Culture Consumables | Pre-warmed complete growth medium, Sterile cryogenic vials, Cell culture vessels [7] [101] | Provide optimal environment for cell recovery post-thaw; proper medium temperature is critical for osmotic balance |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Thawing Optimization Research
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Synth-a-Freeze Cryopreservation Medium | Chemically defined, protein-free cryopreservation medium with 10% DMSO [7] | Suitable for cryopreservation of stem and primary cells; reduces batch-to-batch variability |
| Recovery Cell Culture Freezing Medium | Ready-to-use complete cryopreservation medium with optimized serum ratio [7] | Designed to improve cell viability and recovery after thawing for general mammalian cell culture |
| Dulbecco's Phosphate Buffered Saline (DPBS) | Balanced salt solution without calcium, magnesium, or phenol red [7] | Used for cell washing and dilution; absence of Ca2+/Mg2+ prevents cell clumping |
| Trehalose | Non-permeating cryoprotectant [6] | Protects membranes and proteins by water replacement mechanism; effective in freeze-drying |
| Ice Recrystallization Inhibitors (IRIs) | Synthetic polymers (e.g., PVA), Antifreeze Proteins [99] [98] | Mitigate damage from transient warming events by inhibiting ice crystal growth [98] |
| TrypLE Express | Animal-origin-free dissociation reagent [7] | Gently detaches adherent cells for passaging after thawing and expansion |
Quantitative Data Analysis and Presentation
The effectiveness of thawing optimization can be quantitatively assessed through multiple parameters. The following data, compiled from published studies, illustrates the dramatic impact of thawing conditions on cell survival and function.
Table 4: Impact of Thawing Conditions on Cell Viability and Recovery
| Cell Type/System | Thawing Condition | Key Outcome Metric | Result | Reference Context |
|---|---|---|---|---|
| General Mammalian Cells | Slow thawing (room temperature) | Intracellular ice recrystallization | Significant cell damage | [101] |
| General Mammalian Cells | Rapid thawing (37°C water bath) | Prevention of recrystallization | High cell viability maintained | [101] |
| Cells with DMSO CPA | Exposure to DMSO at room temperature for 1 hour | Reduction in cell viability | ~10% decrease | [101] |
| Lactobacillus rhamnosus GG | Freezing at -196°C (LN₂) + thawing | Bacterial survival rate | 90.94% | [6] |
| Lactobacillus rhamnosus GG | Freeze-drying + suboptimal thawing | Bacterial survival rate | As low as 2% | [6] |
| Cell Therapy Products | Exposure to Transient Warming Events (TWEs) | Delayed Onset Cell Death (DOCD) | Significant apoptosis days post-thaw | [98] |
| Red Blood Cells (freeze-dried) | Cooling rate of 4200 K/min before drying | Cell recovery post-rehydration | Maximal recovery | [102] |
Diagram 2: Thawing Parameter Impact on Outcomes
Optimizing the thawing process to prevent devitrification and recrystallization is a critical component in the broader study of mechanical and osmotic effects of freezing on cells. By understanding the fundamental physical and biological stresses involved—including ice crystal dynamics, osmotic imbalances, and ionic effects—researchers can implement protocols that significantly improve post-thaw cell viability and functionality. The standardized protocols and experimental frameworks presented here provide a foundation for consistent, reproducible results in both research and therapeutic applications. As cryopreservation continues to enable advancements in cell therapy, regenerative medicine, and fundamental biological research, meticulous attention to the thawing process will remain essential for achieving reliable outcomes and maintaining the integrity of precious cellular materials.
Assessing Preservation Efficacy: Validation Methods and Comparative Analysis of Cryopreservation Techniques
Within the broader research on the mechanical and osmotic effects of freezing on cells, post-thaw assessment serves as a critical diagnostic tool. The process of cryopreservation subjects cells to severe physicochemical stresses, including intracellular ice formation, osmotic shock, and solute-induced toxicity, which collectively compromise cellular integrity and function [103] [4]. A comprehensive post-thaw assessment strategy is therefore indispensable for evaluating the success of cryopreservation protocols and for diagnosing the specific points of failure. By quantifying recovery, viability, and function, these methods provide the necessary feedback to refine cryoprotectant formulations, cooling rates, and thawing procedures, ultimately advancing the application of cryopreserved cells in regenerative medicine, assisted reproduction, and biobanking.
This technical guide provides an in-depth overview of current methodologies for post-thaw evaluation, with a specific focus on bridging the gap between observed cell damage and its underlying mechanical and osmotic causes. It details established and emerging techniques for assessing viability, membrane integrity, and cellular function, and provides structured protocols and reagent toolkits to facilitate implementation in the research laboratory.
Core Post-Thaw Assessment Methodologies
A multi-parametric approach is essential for a complete picture of post-thaw cell health. The following table summarizes the key methodologies, their applications, and the specific parameters they measure.
Table 1: Core Post-Thaw Assessment Methodologies
| Assessment Category | Methodology | Measured Parameter(s) | Typical Application & Notes |
|---|---|---|---|
| Viability & Membrane Integrity | SYBR-14/PI Live-Dead Assay [103] | Membrane integrity (viability %) | Differentiates live (SYBR-14+/PI-) from dead (PI+) cells; used in amphibian sperm cryopreservation. |
| Viability & Membrane Integrity | Hypo-Osmotic Swelling Test (HOST) [104] | Membrane functionality and integrity | Assesses sperm membrane integrity by measuring coiled tails in hypo-osmotic medium. |
| Viability & Membrane Integrity | Live, Apoptosis-Negative (LAN) Assay [105] | Viable, non-apoptotic cell count | Flow cytometry-based assay for cord blood mononuclear cells; indicates healthy, functional cells. |
| Viability & Membrane Integrity | Standard Plate Count [6] | Colony Forming Units (CFU) | Determines viable bacterial count (e.g., Lactobacillus) based on reproductive capacity post-thaw. |
| Metabolic & Mitochondrial Function | Computer-Assisted Sperm Analysis (CASA) [104] | Motility %, kinematic parameters (VCL, VSL, LIN) | Objective, quantitative assessment of sperm motility and movement characteristics. |
| Metabolic & Mitochondrial Function | Mitochondrial Membrane Potential (MMP) Assay [106] [104] | Mitochondrial activity and health | Uses JC-1 or similar dyes; high MMP indicates energetic, healthy sperm. |
| Metabolic & Mitochondrial Function | ATP Assay [106] | Intracellular ATP concentration | Direct measure of cellular energy status; critical for functions like sperm motility. |
| Oxidative Stress & Damage | Reactive Oxygen Species (ROS) Assay [106] [104] | Intracellular ROS levels | Elevated ROS indicates oxidative stress, a key cause of post-thaw damage. |
| Oxidative Stress & Damage | Lipid Peroxidation (LPO) Assay [106] [104] | Malondialdehyde (MDA) levels | Measures oxidative damage to cell membranes, a critical failure point in cryopreservation. |
| Oxidative Stress & Damage | Antioxidant Enzyme Activity [104] | SOD, GPx, TAC | Assesses the cellular antioxidant defense system post-thaw. |
Detailed Experimental Protocols
Membrane Integrity and Viability Assessment
The SYBR-14/PI Live-Dead assay is a standard for quantifying membrane-intact cells. The following protocol is adapted from studies on endangered amphibian sperm [103].
Protocol: SYBR-14/PI Staining for Sperm Viability
- Reagent Preparation: Obtain a commercial LIVE/DEAD Sperm Viability Kit. Prepare working solutions as per manufacturer instructions.
- Sample Thawing: Thaw frozen sperm samples using the standardized protocol for the cell type (e.g., rapid thaw in a 37°C water bath for 30 seconds).
- Staining: Aliquot a known volume of thawed sperm suspension (e.g., 100 µL). Add 5 µL of SYBR-14 working solution and incubate for 5-10 minutes at room temperature protected from light.
- Counterstaining: Add 5 µL of propidium iodide (PI) working solution to the same tube and incubate for an additional 5 minutes protected from light.
- Analysis: Place a 5-10 µL aliquot on a microscope slide, cover with a coverslip, and immediately examine under an epifluorescence microscope using appropriate filter sets.
- SYBR-14 (Live): Fluoresces green (emission ~515 nm) in membrane-intact cells.
- Propidium Iodide (Dead): Fluoresces red (emission ~617 nm) in cells with compromised membranes.
- Quantification: Count a minimum of 200 cells across multiple random fields. Calculate viability as: % Viability = (Number of green-fluorescent cells / Total cells counted) × 100.
Functional Motility Assessment
Computer-Assisted Sperm Analysis (CASA) provides robust, objective data on sperm motility. The protocol below is derived from poultry sperm analysis [104].
Protocol: CASA for Sperm Motility
- Instrument Calibration: Calibrate the CASA system (e.g., Hamilton Thorne CASA) according to manufacturer specifications prior to use. Standardize settings for the specific species/cell type.
- Sample Preparation: Thaw sperm samples and dilute 1:10 with an appropriate isotonic extender buffer (e.g., Lake buffer) to achieve an optimal concentration for analysis (~20-40 million cells/mL).
- Loading: Pipette 3-5 µL of the diluted sample onto a pre-warmed (37°C) counting chamber (e.g., Leja slide).
- Data Acquisition: Place the chamber on the preheated stage of the CASA microscope. Acquire data from a minimum of five random fields, capturing at least 200 cells per sample. Standardize acquisition settings (e.g., frame rate: 60 Hz, number of frames: 30).
- Parameter Analysis: The CASA software automatically analyzes and outputs key parameters, including:
- Total Motility (%): Percentage of sperm with any movement.
- Progressive Motility (%): Percentage of sperm moving actively along a straight or circular path.
- Kinematics: Curvilinear Velocity (VCL, µm/s), Straight-Line Velocity (VSL, µm/s), Average Path Velocity (VAP, µm/s), Linearity (LIN = VSL/VCL × 100), and Amplitude of Lateral Head Displacement (ALH, µm).
Assessment of Oxidative Stress
Measuring reactive oxygen species (ROS) is critical for diagnosing oxidative damage. The following fluorometric assay is used for sperm and other cell types [106] [104].
Protocol: Intracellular ROS Assay
- Reagent: Prepare a stock solution of a cell-permeant ROS-sensitive fluorescent probe, such as 2',7'-Dichlorodihydrofluorescein diacetate (H2DCFDA) or Dihydroethidium (DHE), in DMSO.
- Staining: Wash the post-thaw cell pellet with a clean buffer to remove extracellular cryoprotectants. Resuspend the cell pellet at a concentration of ~1-2 million cells/mL in buffer. Add H2DCFDA to a final concentration of 5-10 µM and incubate for 30-45 minutes at 37°C in the dark.
- Washing and Analysis: Centrifuge the cells to remove excess dye and resuspend in fresh buffer.
- Flow Cytometry: Analyze the fluorescence intensity of the cell suspension immediately using a flow cytometer with an excitation wavelength of 488 nm and emission detection at 530 nm. Analyze at least 10,000 events per sample.
- Fluorometry/Plate Reader: Transfer the stained suspension to a black-walled 96-well plate and measure fluorescence.
- Quantification: Express results as Mean Fluorescence Intensity (MFI) relative to an unstained control or a control group (e.g., fresh cells). Higher fluorescence indicates higher levels of intracellular ROS.
Visualizing Cryoinjury Mechanisms and Assessment Pathways
The following diagram integrates the mechanisms of cryopreservation damage with the corresponding assessment methods, creating a direct link between cause and effect that guides post-thaw analysis.
Diagram 1: A diagnostic map linking primary cryoinjury mechanisms to specific types of cellular damage and the corresponding post-thaw assessment methods used to detect them. For example, oxidative stress directly causes lipid peroxidation (LPO), which is quantified using an LPO assay. Similarly, damage to organelles like mitochondria is probed using mitochondrial membrane potential (MMP) and ATP assays.
The Scientist's Toolkit: Key Reagents and Materials
Successful post-thaw assessment relies on a suite of specialized reagents and instruments. The following table details essential items for establishing these assays in the laboratory.
Table 2: Essential Research Reagent Solutions for Post-Thaw Assessment
| Category/Item | Specific Examples | Function & Application |
|---|---|---|
| Viability Stains | SYBR-14, Propidium Iodide (PI) [103] | Fluorescent live/dead staining for membrane integrity. |
| Viability Stains | LIVE/DEAD Fixable Viability Dyes | For flow cytometry; allows cell fixation after staining. |
| Metabolic Probes | JC-1, Tetramethylrhodamine (TMRM) [104] | Assessment of mitochondrial membrane potential. |
| Oxidative Stress Probes | H2DCFDA, Dihydroethidium (DHE) [104] | Detection of intracellular reactive oxygen species (ROS). |
| Lipid Peroxidation Assay Kits | Thiobarbituric Acid Reactive Substances (TBARS) Assay Kits [106] | Quantification of malondialdehyde (MDA), a marker for oxidative membrane damage. |
| Antioxidant Assay Kits | Superoxide Dismutase (SOD), Glutathione Peroxidase (GPx) Kits [104] | Measurement of antioxidant enzyme activity. |
| Specialized Media & Buffers | Lake Extender Buffer [104] | A defined medium for diluting and cryopreserving avian sperm. |
| Specialized Media & Buffers | Hypo-Osmotic Swelling Test Solution [104] | A fructose/sodium citrate solution of defined osmolality (~100 mOsm) for testing sperm membrane functionality. |
| Specialized Media & Buffers | OncoPro Tumoroid Culture Medium [107] | Medium designed for the culture and cryopreservation of 3D patient-derived tumoroids. |
| Cell Processing Kits | EasySep Direct Human PBMC Isolation Kit [105] | Magnetic bead-based kit for isolating mononuclear cells from complex samples like cord blood post-thaw. |
| Instrumentation | Computer-Assisted Sperm Analyzer (CASA) [104] | Automated system for objective analysis of sperm motility and kinematics. |
| Instrumentation | Flow Cytometer | Essential for high-throughput, multi-parameter analysis of viability, apoptosis, ROS, and mitochondrial function. |
| Instrumentation | Controlled-Rate Freezer [4] | Provides reproducible, linear cooling rates critical for optimizing cryopreservation protocols. |
A comprehensive and multi-faceted approach to post-thaw assessment is fundamental to advancing the science of cryopreservation. By systematically applying the methodologies outlined in this guide—from basic membrane integrity tests to functional metabolic and oxidative stress assays—researchers can move beyond simple viability counts to gain a deep, mechanistic understanding of cryoinjury. This detailed profiling is indispensable for reverse-engineering the osmotic and mechanical stresses incurred during freezing and for developing targeted strategies to mitigate them. As the field progresses towards more complex cell types and tissue-like constructs, the integration of these robust assessment protocols will be crucial for ensuring the functional fidelity of cryopreserved biological materials in clinical and biotechnological applications.
Comparative Analysis of Slow Freezing Versus Vitrification Outcomes
Cryopreservation technologies represent a critical component of modern biomedical research, clinical practice, and biobanking. This comprehensive analysis examines the comparative outcomes of slow freezing versus vitrification, two principal cryopreservation methods with distinct thermodynamic pathways and biological consequences. Through systematic evaluation of current literature and experimental data, we demonstrate that methodological refinements in both approaches continue to narrow the efficacy gap, with vitrification generally superior for stress-sensitive specimens like oocytes and complex tissues, while optimized slow freezing protocols remain viable for many cell types. The selection between these methods necessitates careful consideration of specimen characteristics, intended applications, and practical laboratory constraints, with ongoing research focusing on minimizing cryoinjury through improved cryoprotectant formulations and precise thermal control.
Cryopreservation enables the long-term storage of biological specimens by reducing temperatures to levels that dramatically suppress biochemical activity and cellular metabolism [62]. The two dominant methodologies—slow freezing and vitrification—follow distinct thermodynamic paths that impose different mechanical and osmotic stresses on cellular structures [62]. Slow freezing involves controlled, gradual cooling typically at approximately -1°C/minute, facilitating cellular dehydration before intracellular ice formation occurs [75]. In contrast, vitrification achieves an amorphous, glass-like state through ultra-rapid cooling with high cryoprotectant concentrations, thereby avoiding ice crystallization entirely [62] [108]. Understanding the comparative outcomes of these approaches is essential for optimizing preservation protocols across diverse biological systems, from individual cells to complex tissues.
The fundamental physical processes underlying these methods dictate their specific applications and limitations. During slow freezing, extracellular ice formation initiates a solute concentration gradient that drives osmotic water efflux from cells, promoting protective dehydration but risking excessive volumetric changes and solute effects [62]. Vitrification circumvents ice formation but requires potentially toxic concentrations of cryoprotectants and extremely rapid cooling and warming rates to prevent devitrification [62] [108]. This technical review examines the mechanistic basis of both methods, their comparative effectiveness across biological systems, and recent advances that enhance their utility in research and clinical contexts.
Thermodynamic Principles and Cryoinjury Mechanisms
Phase Transition Pathways
The thermodynamic pathways of slow freezing and vitrification diverge significantly in their approach to managing the water-ice transition. Slow freezing follows the path A→C→E→F→G→I→L→Z, where ice crystals initiate at point E and propagate through freeze concentration (E→F→G), progressively increasing extracellular osmolality and driving cellular dehydration [62]. Conventional vitrification follows path A→D→II→M→Z, employing high-concentration cryoprotectants at non-freezing temperatures followed by rapid cooling to achieve a glassy state [62]. Low-CPA vitrification attempts to balance these approaches with path A→C→III→N→Z, utilizing extremely high cooling rates to reduce cryoprotectant requirements [62].
The physical behavior of water and solutes during these processes directly influences cell viability. Ice formation during slow freezing creates mechanical stresses that can disrupt plasma membranes and subcellular structures, while the concomitant solute concentration effects can denature proteins and alter membrane properties [62]. Vitrification avoids ice crystallization but subjects cells to substantial osmotic stresses during cryoprotectant loading and unloading, plus potential chemical toxicity from high cryoprotectant concentrations [108].
Intracellular Ice Formation and Recrystallization
Intracellular ice formation represents a particularly lethal event during cryopreservation, almost invariably resulting in cell death due to irreversible damage to organelles and membrane systems [62]. Advanced modeling approaches now incorporate not only ice nucleation and growth during cooling but also recrystallization during warming, providing more accurate predictions of cryoinjury [13]. These models simulate coupled transport of water and permeable cryoprotectants across cell membranes, describing how intracellular ice volume evolves throughout the freeze-thaw cycle [13].
The following diagram illustrates the key cellular responses and injury mechanisms associated with different temperature profiles during cryopreservation:
Comparative Performance Across Biological Systems
Reproductive Cells and Embryos
Human oocytes demonstrate markedly different outcomes depending on cryopreservation methodology. A 2025 study comparing modified slow freezing with vitrification revealed that while traditional slow freezing with conventional rehydration achieved only 65.1% survival, a modified rehydration protocol dramatically improved survival to 89.8%—statistically equivalent to the 89.7% survival rate with vitrification [109] [110]. Clinical outcomes reflected this improvement, with modified slow freezing achieving pregnancy and implantation rates of 33.8% and 25.5% respectively, comparable to vitrification outcomes of 30.1% and 26.6% [109] [110]. These findings challenge the prevailing consensus that vitrification is invariably superior for oocyte cryopreservation.
For cleavage-stage embryos, vitrification demonstrates clear advantages over conventional slow freezing. A comprehensive comparison of 485 slow-frozen embryos versus 502 vitrified embryos revealed significantly better outcomes with vitrification across all measured parameters [111]. Post-thaw survival rates were 96.95% with vitrification versus 69.06% with slow freezing, while the percentage of embryos retaining excellent morphology was 94.17% versus 60.8% [111]. These morphological advantages translated to superior clinical outcomes, with vitrification yielding higher clinical pregnancy rates (41.53% vs. 21.53%) and implantation rates (14.41% vs. 7.01%) [111].
Table 1: Comparative Outcomes for Human Oocytes and Embryos
| Specimen Type | Method | Survival Rate | Pregnancy Rate | Implantation Rate | Key Findings |
|---|---|---|---|---|---|
| Human oocytes [109] [110] | Traditional slow freezing | 65.1% | 23.5% | 13.8% | Suboptimal outcomes with conventional protocols |
| Modified slow freezing | 89.8% | 33.8% | 25.5% | Improved rehydration protocol closed gap with vitrification | |
| Vitrification | 89.7% | 30.1% | 26.6% | Considered gold standard for oocyte cryopreservation | |
| Cleavage-stage embryos (Day 3) [111] | Slow freezing | 69.06% | 21.53% | 7.01% | Significantly inferior outcomes across all parameters |
| Vitrification | 96.95% | 41.53% | 14.41% | Superior survival, morphology, and clinical results |
Ovarian and Testicular Tissues
Complex tissues present additional cryopreservation challenges due to their heterogeneous cellular composition and three-dimensional architecture. For ovarian tissue, a 2024 transplantation study compared two vitrification protocols (VF1, VF2) against slow freezing (SF) in a nude mouse model [112]. While all methods supported restoration of endocrine function post-transplantation, the VF2 vitrification protocol yielded significantly higher estradiol levels at 6 weeks post-transplantation compared to slow freezing [112]. Histological assessment revealed a higher proportion of normal follicles in vitrification groups, with VF2 demonstrating significantly better preservation compared to slow freezing at the 6-week endpoint [112].
For testicular tissue, both slow freezing and vitrification present distinct advantages and limitations. Slow freezing utilizes lower cryoprotectant concentrations and controlled cooling to minimize intracellular ice formation, but risks freeze-induced injury and requires expensive equipment [108]. Vitrification offers faster processing with reduced ice crystal formation but demands technical expertise and introduces potential cytotoxicity from high cryoprotectant concentrations [108]. Sample size emerges as a critical factor, with optimal testicular tissue fragments ranging from 0.3 to 1.5 mm³ to balance cryoprotectant penetration and heat transfer [108].
Table 2: Tissue Cryopreservation Outcomes Following Transplantation
| Tissue Type | Method | Follicular Integrity | Endocrine Function | Stromal Apoptosis | Angiogenesis |
|---|---|---|---|---|---|
| Ovarian tissue [112] | Slow freezing (SF) | Lower normal follicle rate at 6 weeks | Lower estradiol levels | Higher apoptosis at 4 weeks | Better CD31 expression |
| Vitrification (VF1) | Intermediate preservation | Intermediate recovery | Lower apoptosis | Reduced angiogenesis | |
| Vitrification (VF2) | Best preservation at 6 weeks | Highest estradiol levels | Lower apoptosis | Intermediate results | |
| Testicular tissue [108] | Slow freezing | Maintains tissue architecture | N/A | Risk of freeze-induced injury | Preserves cell interactions |
| Vitrification | Better cell survival | N/A | Reduced ice crystal damage | Requires technical expertise |
Experimental Protocols and Methodologies
Vitrification Protocols for Ovarian Tissue
VF1 Protocol (Based on Amorim et al. with modifications) [112]:
- Equilibration: 3.8% ethylene glycol (EG), 0.5M sucrose, 6% Serum Substitute Supplement (SSS) in MEM-Glumax for 3 minutes at room temperature
- Transition: Transfer to 19% EG, 0.5M sucrose in MEM-Glumax with 6% SSS for 1 minute
- Vitrification: 38% EG, 0.5M sucrose, MEM-Glumax with 6% SSS for 11 minutes
- Cooling: Placement on metallic grid followed by plunging into liquid nitrogen
- Warning: Sequential incubation in decreasing sucrose concentrations (0.5M, 0.25M, 0.125M, 0M) with 6% SSS for 5 minutes each at room temperature
VF2 Protocol (Based on Kagawa et al. with modifications) [112]:
- First step: 10% EG, 10% DMSO, 20% SSS in M199 for 25 minutes at room temperature
- Vitrification solution: 20% EG, 20% DMSO, 0.5M sucrose, 20% SSS in M199 for 15 minutes at room temperature
- Thawing: 1M sucrose, 20% SSS in M199 at 37°C for 1 minute, followed by 0.5M, 0M, 0M sucrose solutions with 20% SSS for 5 minutes each
Slow Freezing Protocol for Ovarian Tissue
SF Protocol (Based on vonWolff et al. with modifications) [112]:
- Freezing medium: Leibovitz L-15 medium with 10% SSS, 10% DMSO, 0.1M sucrose
- Equilibration: 30 minutes at 4°C
- Cooling program:
- From 2°C to -6°C at 2°C/minute
- Manual ice seeding
- From -6°C to -40°C at 0.3°C/minute
- From -40°C to -140°C at 10°C/minute
- Transfer to liquid nitrogen for storage
- Thawing: 37°C water bath for 2 minutes, followed by stepwise dilution in L-15 medium with decreasing DMSO and sucrose concentrations
General Cell Freezing Protocol
A standardized approach for cell culture cryopreservation includes [7] [75]:
- Harvesting: Collect log-phase cells at >80% confluency with >90% viability
- Cryoprotectant medium: Complete growth medium with 10% DMSO or specialized commercial freezing media
- Cooling rate: Approximately -1°C/minute using controlled-rate freezer or isopropanol containers
- Storage: Transfer to liquid nitrogen vapor phase (-135°C to -196°C) for long-term preservation
The following workflow diagram illustrates the key decision points and procedures for selecting and implementing appropriate cryopreservation methodologies:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Equipment for Cryopreservation Research
| Category | Specific Products/Components | Function and Application |
|---|---|---|
| Cryoprotectants | Dimethyl sulfoxide (DMSO), Ethylene glycol (EG), 1,2-Propanediol (PrOH) | Penetrating agents that reduce ice formation and stabilize membranes [7] [112] |
| Non-penetrating agents | Sucrose, trehalose, human serum albumin (HSA) | Osmotic buffers that control cell volume changes during CPA addition/removal [47] [112] |
| Commercial freezing media | CryoStor CS10, Synth-a-Freeze, Recovery Cell Culture Freezing Medium | Optimized, ready-to-use formulations for specific cell types [7] [75] |
| Specialized media | mFreSR (for hES/iPS cells), MesenCult-ACF (for MSCs) | Cell-type specific formulations preserving differentiation potential [75] |
| Cooling devices | Controlled-rate freezers, Mr. Frosty, CoolCell | Achieve standardized cooling rates (~1°C/min) for reproducible results [7] [75] |
| Storage systems | Cryogenic vials, liquid nitrogen tanks | Maintain temperatures below -135°C for long-term stability [7] [75] |
| Assessment tools | Differential scanning calorimetry, viability stains, membrane integrity assays | Characterize thermodynamic properties and evaluate post-thaw recovery [13] [47] |
The comparative analysis of slow freezing versus vitrification outcomes reveals a nuanced technological landscape where method selection must be guided by specimen characteristics, intended applications, and practical laboratory considerations. Vitrification generally demonstrates superior performance for stress-sensitive specimens like oocytes and early embryos, particularly where maximum cellular viability is essential. However, recent methodological refinements to slow freezing protocols—particularly improved rehydration methods—have substantially narrowed this performance gap, making optimized slow freezing a viable alternative for many applications.
Future advancements in cryopreservation methodology will likely focus on reducing both mechanical and osmotic injuries through improved cryoprotectant formulations, precise thermal control, and specimen-specific protocol optimization. The integration of advanced modeling approaches that predict intracellular ice formation and recrystallization represents a promising direction for rational protocol design rather than empirical optimization. As cryopreservation technologies continue to evolve, both slow freezing and vitrification will maintain important roles in the preservation of biological systems for research, clinical application, and biodiversity conservation.
The study of the mechanical and osmotic effects of freezing on cells is a critical area of research in cryobiology, with profound implications for cell therapy, tissue engineering, and fundamental biological science. When cells undergo freezing, they face two primary classes of injury: osmotic stress due to solute concentration and water efflux, and mechanical damage from ice crystal formation [73] [2]. Understanding these complex phenomena requires sophisticated analytical techniques that can probe cellular responses at molecular, structural, and temporal scales. Cryomicroscopy, calorimetry, and Raman spectroscopy have emerged as powerful, complementary tools for investigating these freezing injury mechanisms, each providing unique insights into the biophysical processes that determine cell survival during cryopreservation.
The osmotic stress hypothesis posits that as extracellular ice forms, unfrozen channels become constricted, and solute concentration rises, leading to cellular dehydration and volumetric changes that can exceed tolerable limits [73] [2]. Conversely, the mechanical injury hypothesis suggests that intracellular ice formation (IIF) physically disrupts cellular structures, often proving fatal to cells [113]. Advanced analytical techniques enable researchers to not only observe these phenomena but also quantify them, leading to improved cryopreservation protocols that maximize cell viability and function post-thaw. This technical guide explores the principles, methodologies, and applications of these key techniques within the context of freezing injury research.
Theoretical Framework: Mechanical and Osmotic Injury Mechanisms
Osmotic Stress as a Mechanism of Freezing Injury
The osmotic mechanism of freezing injury occurs during slow cooling, where ice forms initially in the extracellular solution. This extracellular ice formation increases the solute concentration in the remaining unfrozen fraction, creating a chemical potential difference that drives water out of cells. The consequent cellular dehydration and shrinkage can cause injury if volumetric excursions exceed tolerable limits [73]. A key concept is the "unfrozen channel" size hypothesis, which suggests that injury during slow freezing correlates more strongly with the reduction in the size of these channels than with the absolute solute concentration [2]. This dehydration can potentially lead to membrane rupture due to osmotic pressure gradients, allowing extracellular ice to propagate into the cytoplasm—a concept formalized as the osmotic rupture hypothesis of intracellular freezing injury [113].
Table 1: Key Mechanisms of Freezing Injury
| Injury Mechanism | Conditions | Primary Cause | Cellular Manifestation |
|---|---|---|---|
| Slow-Freezing Injury (Osmotic) | Slow cooling rates (<1°C/min) | Solution effects, solute concentration, cell dehydration | Excessive shrinkage, membrane damage, loss of function |
| Intracellular Ice Formation (Mechanical) | Rapid cooling rates (>50°C/min) | Incomplete dehydration, supercooling | Physical disruption of organelles and membranes |
| Osmotic Shock | During addition/removal of cryoprotectants | Rapid volumetric changes | Membrane rupture, lysis |
| Cryoprotectant Toxicity | Prolonged exposure pre-freeze or post-thaw | Chemical effects of cryoprotectants | Metabolic alterations, cytoskeleton changes |
Intracellular Ice Formation and Mechanical Damage
When cooling is too rapid, insufficient time exists for cellular dehydration to eliminate supercooling, resulting in intracellular ice formation (IIF). Equations have been developed that describe the kinetics of water loss and predict the likelihood of IIF as a function of cooling rate, with predictions agreeing well with experimental observations [2]. Although avoiding IIF is typically necessary for survival, it is not sufficient, as slow freezing itself can be injurious [2]. The osmotic rupture hypothesis provides a mechanistic link between these two damage pathways, suggesting that osmotically driven water efflux during freezing produces sufficient pressure to rupture the plasma membrane, thereby allowing extracellular ice to propagate into the cytoplasm [113].
Diagram 1: Freezing injury mechanisms and pathways.
Cryomicroscopy: Visualizing Freezing Processes in Real-Time
Principles and Instrumentation
Cryomicroscopy combines conventional microscopy with precisely controlled freezing stages, enabling direct observation of cellular responses during freezing and thawing. This technique allows researchers to visualize intracellular ice formation, cellular deformation, and osmotic responses in real-time [114]. Modern cryomicroscopy systems incorporate high-speed cameras, temperature-controlled stages capable of precise cooling rate control (typically from 0.1°C/min to 100°C/min), and specialized software for image analysis. The temperature range typically spans from +37°C to -150°C, covering the critical phase transition zones where most freezing-related injuries occur. Advanced systems now incorporate time-deterministic cryo-optical microscopy that can rapidly freeze biological samples in milliseconds during observation, capturing dynamic cellular processes with high temporal resolution [115].
Experimental Protocols for Cryomicroscopy
Sample Preparation:
- Cells are typically suspended in appropriate medium with or without cryoprotective agents.
- For adherent cells, special coverslips compatible with the cryostage are used.
- Sample volume is minimized to ensure rapid and uniform heat transfer during freezing.
- Fluorescent dyes (e.g., calcium indicators, membrane integrity probes) may be incorporated for functional imaging [115].
Freezing Protocol:
- Place sample on temperature-controlled stage and stabilize at initial temperature (typically 20-37°C).
- Initiate cooling at predetermined rate (e.g., 1°C/min to 50°C/min) while recording images.
- For rapid freezing experiments, introduce liquid cryogen (e.g., propane-isopentane mixture) at precisely controlled timing [115].
- Continue imaging through the phase transition temperatures and until target temperature is reached.
- For thawing experiments, initiate warming at controlled rates (typically >60°C/min).
Data Analysis:
- Quantify intracellular ice formation events by detecting sudden darkening or flashing within cells.
- Measure cell volume changes by tracking membrane boundaries over time.
- Calculate the fraction of cells exhibiting IIF at different cooling rates.
- Correlate freezing events with temperature profiles to determine nucleation temperatures.
Table 2: Cryomicroscopy Applications in Freezing Injury Research
| Application | Measured Parameters | Technical Requirements | Key Insights |
|---|---|---|---|
| Intracellular Ice Detection | IIF temperature, incidence, morphology | High-speed imaging (>100 fps) | Correlation between cooling rate and IIF probability |
| Osmotic Response Analysis | Cell volume changes, kinetics | High contrast optics, membrane labels | Membrane permeability parameters (Lp, Ea) |
| Cryoprotectant Screening | IIF suppression, toxicity effects | Multi-well capability, viability staining | Optimal CPA type and concentration |
| Ice Crystal Morphology | Extracellular ice structure, channel size | Polarized light optics | Relationship between ice structure and cell damage |
Calorimetry: Quantifying Thermal Transitions During Freezing
Principles and Instrumentation
Calorimetry provides quantitative information on heat flows and thermal transitions during freezing processes. Differential Scanning Calorimetry (DSC) is the primary calorimetric technique used in cryobiology, measuring the heat released or absorbed during phase transitions as a function of temperature or time. DSC instruments consist of matched sample and reference cells with precise temperature control, detecting the differential heat flow required to maintain both cells at the same temperature during programmed cooling or warming. Modern DSC systems can operate at cooling rates relevant to cryopreservation (0.1°C/min to 100°C/min) and detect subtle thermal events associated with intracellular freezing, devitrification, and crystallization of cryoprotective solutions.
Experimental Protocols for Cryopreservation Calorimetry
Sample Preparation:
- Prepare cell suspensions at standardized concentrations (typically 10⁶-10⁷ cells/mL).
- Incorporate cryoprotective agents at desired concentrations.
- Use small sample volumes (5-50 μL) to ensure temperature uniformity.
- Include appropriate reference solutions (medium without cells) for baseline subtraction.
Freezing Protocol:
- Load sample and reference into DSC pans and seal properly.
- Equilibrate at initial temperature (e.g., 25°C).
- Program cooling protocol to match cryopreservation conditions of interest.
- Typical cooling rates range from 1°C/min to 50°C/min, extending to target temperature (e.g., -100°C).
- For warming experiments, program appropriate heating rates (typically 5-20°C/min).
Data Analysis:
- Identify thermal events (ice crystallization, melting, devitrification) as peaks in the heat flow curve.
- Integrate peak areas to determine enthalpy changes (ΔH) for phase transitions.
- Calculate the amount of unfrozen water at different temperatures.
- Determine homogeneous nucleation temperatures from exothermic events.
- Analyze glass transition temperatures from changes in heat capacity.
Raman Spectroscopy: Molecular-Level Analysis of Frozen Cells
Principles and Instrumentation
Raman spectroscopy is a label-free, non-destructive technique that provides molecular-level information based on inelastic scattering of monochromatic light. When applied to cryopreservation research, it enables detection of biochemical changes, intracellular ice formation, and cryoprotectant distribution in cells and tissues [116] [114]. The technique measures vibrational modes of molecular bonds, generating spectral fingerprints that are specific to different cellular components including proteins, lipids, nucleic acids, and ice crystals. Raman cryomicroscopy combines Raman spectroscopy with low-temperature microscopy, allowing hyperspectral imaging of frozen samples while preserving their physicochemical states [116] [114]. Recent advances have demonstrated that cryofixation enables long exposure times under stabilized low-temperature conditions, significantly improving signal-to-noise ratio and spatial resolution in Raman imaging [116].
Experimental Protocols for Raman Cryomicroscopy
Sample Preparation:
- Cells are suspended in appropriate medium with or without cryoprotectants.
- For single-cell analysis, cells are typically adhered to fused silica coverslips compatible with Raman measurements.
- Sample volume is minimized to facilitate rapid freezing.
- For some applications, Raman tags (e.g., alkyne-tagged molecules) may be incorporated to track specific molecules [116].
Freezing and Measurement Protocol:
- Place sample on custom cryostat stage capable of maintaining stable low temperatures (e.g., 233 K) [116].
- For rapid freezing, apply liquid cryogen (e.g., propane at 88 K) to avoid destruction of cell structures by large ice crystal formation [116].
- Stabilize sample temperature for Raman measurement using liquid nitrogen circulation system.
- Acquire Raman spectra using laser excitation appropriate for the sample (typically 532 nm or 785 nm).
- For imaging, perform hyperspectral mapping with typical acquisition times of 0.1-10 seconds per spectrum.
- Maintain consistent laser power to avoid sample heating or degradation.
Data Analysis:
- Pre-process spectra (background subtraction, cosmic ray removal, normalization).
- Identify characteristic Raman bands: O-H stretching (~3130 cm⁻¹ for ice), C-H stretching (2850-2930 cm⁻¹ for lipids), amide I (1660-1680 cm⁻¹ for proteins), and cytochrome bands (750, 917, 968, 1337 cm⁻¹) [116].
- Use multivariate analysis (PCA, cluster analysis) to identify spectral patterns correlated with freezing injury.
- Generate chemical images based on specific band intensities to visualize spatial distribution of components.
Diagram 2: Raman cryomicroscopy workflow for cell analysis.
Key Raman Spectral Signatures in Frozen Cells
Raman spectroscopy detects distinct molecular signatures that change during freezing processes. The following key spectral features provide insights into freezing injury mechanisms:
- Ice Detection: The O-H stretching band shifts and narrows upon ice formation, typically observed around 3130 cm⁻¹, allowing discrimination between liquid water and ice phases within samples [116].
- Lipid Phase Changes: Raman bands at 1061 cm⁻¹ (C-C stretching) and 2880 cm⁻¹ (asymmetric CH₂ stretching) show intensity increases at low temperatures (233 K), indicating lipid phase transitions to more ordered states [116].
- Protein Conformation: The amide I band (1680 cm⁻¹) and amide III region provide information on protein secondary structure changes that may occur during freezing.
- Cytochrome States: Bands at 750, 917, 968, and 1337 cm⁻¹ provide information on cytochrome redox states, which can be preserved by cryofixation and indicate metabolic status at the moment of freezing [116].
- Resonance Raman Signals: Carotenoid bands at 1153 and 1517 cm⁻¹ appear enhanced at low temperatures, enabling localization of lipid droplets without additional labeling [116].
Table 3: Key Raman Bands for Analyzing Frozen Cells
| Raman Shift (cm⁻¹) | Assignment | Interpretation in Frozen Cells | Application in Freezing Research |
|---|---|---|---|
| 3130 | O-H stretching (ice) | Water-to-ice phase transition | Detection of intracellular ice formation |
| 2850 | CH₂ symmetric stretch (lipids) | Lipid packing and ordering | Membrane phase transitions at low temperature |
| 2880 | CH₂ asymmetric stretch (lipids) | Lipid acyl chain order | Detection of lipid phase changes |
| 1660-1680 | Amide I (proteins) | Protein secondary structure | Protein denaturation assessment |
| 750, 968 | Cytochromes | Heme protein redox state | Metabolic status preservation by cryofixation |
| 1153, 1517 | Carotenoids | Vitamin storage in lipid droplets | Localization of hydrophobic compartments |
Integrated Experimental Approaches and Applications
Correlative Microscopy and Spectroscopy
The most powerful insights into freezing injury mechanisms often come from integrating multiple analytical techniques. Correlative approaches combine the spatial and temporal resolution of cryomicroscopy with the molecular specificity of Raman spectroscopy and the quantitative thermal data from calorimetry. For example, researchers can use cryomicroscopy to identify the temperature of intracellular ice formation in specific cells, then employ Raman spectroscopy to analyze the biochemical composition of those same cells, correlating IIF events with molecular signatures. Similarly, DSC data on phase transition temperatures and enthalpies can inform the interpretation of both cryomicroscopy and Raman experiments, creating a comprehensive understanding of the freezing process across multiple scales.
Applications in Cell Therapy and Drug Development
Advanced analytical techniques for studying freezing injury have direct applications in the development and optimization of cell therapies and biopharmaceuticals. As noted, "Off-the-shelf cell therapies have the potential to revolutionize cell and gene therapy," with effective cryopreservation being a key bottleneck in their widespread adoption [81]. Understanding and mitigating freezing injury is essential for preserving the viability, potency, and functionality of therapeutic cells, including stem cells, immune cells, and tissue-engineered products. These techniques enable:
- Optimization of cryopreservation protocols for specific cell types by identifying injury mechanisms and protective strategies.
- Screening of novel cryoprotective agents and formulations that minimize osmotic and mechanical stress.
- Quality control assessment of cryopreserved products through molecular and structural analysis.
- Development of safer, DMSO-free cryopreservation methods essential for direct administration of cell therapies [81].
Research Reagent Solutions for Freezing Injury Studies
Table 4: Essential Research Reagents for Freezing Injury Studies
| Reagent Category | Specific Examples | Function in Research | Application Notes |
|---|---|---|---|
| Cryoprotective Agents | Dimethyl sulfoxide (DMSO), glycerol, sucrose | Protect cells from freezing injury by modulating ice formation and osmotic stress | DMSO (5-10%) most common; sucrose used in Raman studies for its minimal interference [81] [114] |
| Fluorescent Probes | Fluo-4 (Ca²⁺ indicator), membrane integrity dyes (PI), actin probes (SPY-555) | Visualize ionic distributions, cell viability, and structural changes during freezing | Calcium indicators preserved by cryofixation enable ion distribution studies [115] |
| Cell Culture Media | Standard cell culture media, specialized freezing media | Maintain cell health during pre-freeze processing and provide base solution for cryoprotectants | Composition affects osmotic response; should be carefully controlled |
| Raman Tags | Alkyne-tagged molecules (EdU), deuterated compounds | Enable tracking of specific molecules via Raman spectroscopy without interfering background | Alkyne tags show distinct Raman peaks in silent spectral regions (2000-2300 cm⁻¹) [116] |
| Cryogens | Liquid nitrogen, propane-isopentane mixtures | Enable rapid freezing for cryofixation with minimal ice crystal artifacts | Propane-isopentane (88K) preferred for rapid freezing in Raman studies [116] |
Advanced analytical techniques including cryomicroscopy, calorimetry, and Raman spectroscopy provide powerful, complementary approaches for investigating the mechanical and osmotic effects of freezing on cells. Cryomicroscopy offers direct visualization of freezing processes in real-time, calorimetry delivers quantitative thermal data on phase transitions, and Raman spectroscopy enables molecular-level analysis of frozen samples without the need for labels or extensive processing. Together, these techniques are revealing the complex interplay between osmotic stress and mechanical damage that determines cell survival during cryopreservation. As these technologies continue to advance—with improvements in temporal resolution, sensitivity, and integration—they will further accelerate the development of optimized cryopreservation protocols for cell therapies, biobanking, and fundamental biological research.
The long-term preservation of cells and microorganisms is a cornerstone of modern biological research, biobanking, and the development of cell-based therapies. Cryopreservation halts biochemical activity, enabling the storage of biological samples for indefinite periods. However, the process of freezing and thawing induces significant mechanical and osmotic stress that can compromise cell viability and function. The formation of intracellular and extracellular ice crystals can cause direct mechanical damage to cellular structures, while the resultant solute imbalances lead to osmotic shock, dehydration, and membrane rupture [75] [117]. The efficacy of cryopreservation is therefore critically dependent on the use of cryoprotective agents (CPAs) designed to mitigate these damaging effects. This whitepaper evaluates the efficacy of various cryoprotectants across a spectrum of biological materials—from mammalian cells to microorganisms—situating the analysis within the core thesis of understanding the mechanical and osmotic consequences of freezing. The selection of an optimal CPA is not a one-size-fits-all endeavor; it must be tailored to the specific biological and structural characteristics of the cell type being preserved [84] [117].
Cryoprotectant Mechanisms and Classifications
Cryoprotectants function through a suite of interconnected mechanisms to protect cells during the freeze-thaw cycle. Understanding these mechanisms is essential for evaluating their efficacy and selecting appropriate formulations.
- Inhibition of Ice Crystal Formation: Permeating CPAs, such as dimethyl sulfoxide (DMSO) and glycerol, depress the freezing point of water and reduce the rate of ice crystal growth. They enter the cell and minimize the formation of lethal intracellular ice [117] [118]. Non-permeating CPAs, such as trehalose and sucrose, remain extracellular, increasing viscosity and promoting vitrification—a glass-like state that prevents ice formation altogether [118].
- Osmotic Regulation: During slow cooling, extracellular ice formation elevates the concentration of solutes outside the cell. This creates an osmotic gradient that draws water out, preventing intracellular ice formation but risking excessive cell dehydration and shrinkage. Permeating CPAs help to balance the osmotic pressure across the membrane, thereby reducing the extent of water efflux and protecting against volumetric injury [117].
- Membrane Stabilization: The phase transition of membrane phospholipids from a liquid crystalline to a gel state during cooling can compromise membrane integrity. Certain cryoprotectants, including some amphiphilic peptides, interact with the lipid bilayer to stabilize it against this phase transition and prevent leakage [117].
- Ice Recrystallization Suppression: During thawing, small ice crystals can fuse to form larger, more damaging ones. Antifreeze proteins (AFPs) and antifreeze peptides (AFpeps) bind to the surface of ice crystals and inhibit this recrystallization process, a mechanism particularly vital for preserving cellular structures [119] [117].
Table 1: Classification and Mechanisms of Common Cryoprotectants
| Cryoprotectant Type | Examples | Permeability | Primary Mechanism of Action |
|---|---|---|---|
| Permeating CPAs | DMSO, Glycerol, Ethylene Glycol | Permeable | Depress freezing point, reduce intracellular ice formation, moderate osmotic shock [117] [118] |
| Non-Permeating CPAs | Trehalose, Sucrose, Polyvinylpyrrolidone (PVP) | Impermeable | Promote extracellular vitrification, induce protective dehydration, inhibit ice recrystallization [118] |
| Macromolecular CPAs | Antifreeze Proteins (AFPs), Antifreeze Peptides (AFpeps), Polymers | Impermeable (typically) | Bind to ice crystals to inhibit growth and recrystallization; some stabilize membranes [119] [117] |
Comparative Efficacy Across Cell Types and Kingdoms
The cryoprotective efficacy of various formulations is highly dependent on the specific cell type, influenced by factors such as cell size, membrane composition, and natural habitat.
Bacteria (Enterobacterales)
A recent study systematically evaluating cryoprotectants for Enterobacterales strains at -20°C demonstrated clear efficacy differences among formulations. The tested cryoprotectants included combinations of permeable agents and nutrient supplements.
Table 2: Efficacy of Different Cryoprotectants for Enterobacterales after 12 Months at -20°C [84]
| Cryoprotectant Formulation | Key Components | Survival Rate (%) |
|---|---|---|
| Cryoprotectant 1 | 70% Glycerin, Peptone, Yeast Extract, Glucose | 88.87% |
| Cryoprotectant 2 | 10% DMSO, 70% Glycerin, Peptone, Yeast Extract, Glucose | 84.85% |
| Cryoprotectant 3 | 10% DMSO, Glucose | 83.50% |
| Cryoprotectant 4 | 70% Glycerin, Glucose | 44.81% |
The superior performance of Cryoprotectant 1 highlights the synergistic benefit of combining a permeable CPA (glycerin) with nutrient supplements (peptone and yeast extract). Notably, the formulation containing only glycerin without nutrients (Cryoprotectant 4) resulted in significantly lower viability, underscoring that CPA selection alone is insufficient; supplementary components are critical for long-term bacterial survival [84].
Yeast (Saccharomyces cerevisiae)
Proteomic analysis of S. cerevisiae revealed that different CPA formulations trigger distinct molecular response pathways. The recovery and viability of yeast post-thaw are directly linked to these proteomic changes. Formulations that induced less stress and maintained proteomic stability corresponded with higher survival rates. This research highlights the move beyond simple viability counts toward understanding the functional molecular mechanisms of cryoprotection [118].
Mammalian Cells (NK Cells and Stem Cells)
The requirements for mammalian cells are often more stringent, with a strong emphasis on preserving not just viability but also specific cellular functions post-thaw.
- Natural Killer (NK) Cells: Research on NK-92 cells shows that exposure to conventional CPAs like DMSO can reduce cell membrane fluidity and cytotoxicity even before freezing. The optimal cooling rate for these cells was identified as 4-5°C/min. The study hypothesized that cell dehydration during freezing disrupts cytolytic granules, causing intracellular damage, and pointed to a need for low-DMSO or DMSO-free solutions that incorporate osmolytes to mitigate functional loss [4].
- Stem Cells: Best practices for cryopreserving sensitive mammalian cells, such as human pluripotent stem cells (ES and iPS cells), involve using specialized, serum-free, defined freezing media like CryoStor CS10 or mFreSR. A controlled cooling rate of approximately -1°C per minute is critical for maximizing post-thaw viability and function [75].
The following diagram illustrates the interconnected osmotic and mechanical stress pathways activated during cell freezing, and the protective mechanisms cryoprotectants use to counter them.
Advanced and Emerging Cryoprotectant Technologies
The field of cryopreservation is evolving beyond traditional CPAs like DMSO and glycerol, driven by the need for reduced toxicity and enhanced functionality.
- Antifreeze Peptides (AFpeps): Derived from natural sources like fish gelatin or designed synthetically, AFpeps represent a promising class of bio-based cryoprotectants. They exhibit dual functionality by simultaneously interacting with ice crystals to suppress their growth and recrystallization, and with proteins (e.g., lactalbumin) to stabilize their structure [119] [117]. Their amphiphilic nature allows some AFpeps to stabilize cell membranes, and they can be engineered to possess additional properties, such as antioxidant or antimicrobial activity, making them multifunctional tools for advanced biopreservation [117].
- Multifunctional Formulations: Research is increasingly focused on developing cocktails that combine permeable and non-permeating CPAs to create synergistic effects. For instance, combining DMSO with trehalose can provide both intracellular and extracellular protection, potentially allowing for a reduction in the concentration of the toxic DMSO while maintaining or improving efficacy [118].
- Supporting Technologies: Techniques like magnetic field-assisted osmotic dehydration have been shown to improve the outcome of freezing for more complex tissues. In model systems like strawberries, this pretreatment results in a more uniform distribution of water, better retention of cellular structure, and reduced damage upon thawing, suggesting potential applications in cellular systems [120].
Experimental Protocols for Efficacy Assessment
A standardized approach to evaluating cryoprotectants is vital for generating comparable data. Below is a generalized protocol that can be adapted for different cell types.
Protocol: Assessing Post-Thaw Viability of Microbial Cultures
This protocol is adapted from methods used in recent studies with Enterobacterales and yeast [84] [118].
- Inoculum Preparation: Grow the microbial culture to its maximum growth phase (log phase). For bacteria, adjust the suspension to a density of 0.5 McFarland units in a phosphate-buffered saline (PBS). For yeast, grow in appropriate broth to an OD600 of ~0.8.
- Cryoprotectant Addition: Concentrate cells via gentle centrifugation. Resuspend the cell pellet in the test cryoprotectant formulations. Common formulations to test include:
- 70% Glycerin with nutrient supplements (e.g., peptone, yeast extract)
- 10% DMSO with 70% Glycerin
- 10% DMSO alone
- Serum-free commercial cryopreservation media
- Aliquoting and Equilibration: Aliquot the cell-cryoprotectant suspension into cryogenic vials. Allow for equilibration at 4-6°C for 30 minutes.
- Controlled-Rate Freezing: Freeze the vials using a controlled-rate freezer. A standard cycle involves cooling at -1°C/min to -40°C, followed by a faster ramp to -80°C or lower. Alternatively, use an isopropanol freezing container placed in a -80°C freezer to achieve an approximate -1°C/min cooling rate.
- Storage: Store vials at the target temperature (e.g., -80°C or in liquid nitrogen vapor) for the desired duration.
- Thawing and Viability Assay: Rapidly thaw vials by agitation in a 37°C water bath for 3-5 minutes. Perform serial dilutions of the thawed suspension and plate onto appropriate solid growth media. Incubate and count colony-forming units (CFU). Calculate the survival rate as a percentage of the pre-freeze viable count.
Protocol: Evaluating Functional Recovery in Mammalian Immune Cells
For therapeutic cells like NK cells, assessing function is as important as viability [4].
- Cell Preparation: Culture and expand the immune cell population (e.g., NK-92 cells) to the required number.
- Cryopreservation: Resuspend cells in test cryoprotectants, which may include low-DMSO formulations or solutions supplemented with osmolytes. Use controlled-rate freezing at an optimized rate (e.g., 4-5°C/min for NK cells).
- Post-Thaw Analysis:
- Viability and Recovery: Use trypan blue exclusion or flow cytometry-based viability dyes to determine immediate post-thaw viability.
- Proliferation Assay: Culture thawed cells and monitor their growth over several days to assess recovery.
- Functional Cytotoxicity Assay: Co-culture thawed NK cells with target cancer cells. Measure specific lysis of targets or quantify the release of cytotoxic granules (perforin, granzyme) to ensure anti-cancer functionality has been retained.
The experimental workflow for a comprehensive cryoprotectant assessment, from cell preparation to data analysis, is outlined below.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful cryopreservation requires a suite of reliable reagents and equipment. The following table details key components of a cryopreservation workflow.
Table 3: Essential Reagents and Materials for Cryopreservation Research
| Item Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Permeating CPAs | Dimethyl Sulfoxide (DMSO), Glycerol | Penetrate cell membrane to protect against intracellular ice formation. DMSO toxicity requires concentration optimization [4] [118]. |
| Non-Permeating CPAs | Trehalose, Sucrose, Polyvinylpyrrolidone (PVP) | Remain outside cell, promote vitrification, inhibit ice recrystallization. Crucial for reducing osmotic stress [118]. |
| Specialized Commercial Media | CryoStor CS10, mFreSR | Ready-to-use, serum-free, defined formulations. Provide consistent, high-performance cryopreservation for specific cell types (e.g., stem cells) [75]. |
| Controlled-Rate Freezing Equipment | Controlled-Rate Freezers, Mr. Frosty, CoolCell | Ensure a consistent, optimal cooling rate (typically ~ -1°C/min), which is critical for high post-thaw viability [75]. |
| Cell Culture Consumables | Cryogenic Vials (e.g., Corning) | Designed for low-temperature storage; use internal-threaded vials to prevent contamination [75]. |
| Analytical Tools | LC-MS/MS instrumentation, Flow Cytometer | Used for deep-dive efficacy assessment (proteomics) and rapid viability/function measurement post-thaw [4] [118]. |
The evaluation of cryoprotectant efficacy is a multifaceted process that must account for the unique biological and physical characteristics of each cell type, from robust microorganisms to therapeutically relevant human cells. The mechanical and osmotic stresses induced by freezing are a universal challenge, but the optimal solution is not universal. As evidenced by the data, formulations combining permeable and non-permeating agents, often supplemented with nutrients or stabilizers, consistently outperform single-component solutions. The future of cryopreservation lies in the development and adoption of advanced, multifunctional cryoprotectants like antifreeze peptides, which offer a mechanism-based approach to protection. Furthermore, integrating supporting technologies and rigorous, standardized experimental protocols that assess both viability and function is paramount for advancing the field. This ensures that cryopreserved cells are not merely alive but are fully functional and ready for use in downstream research and clinical applications.
Validation of Preservation Protocols for Clinical and Biobanking Applications
The expanding fields of regenerative medicine, cell-based therapies, and personalized medicine have increased the critical importance of reliable, validated preservation protocols for biological samples. Within the context of research on the mechanical and osmotic effects of freezing on cells, protocol validation transcends mere sample longevity to encompass the rigorous preservation of cellular function, structural integrity, and biochemical fidelity post-thaw. For clinical and biobanking applications, where samples may determine therapeutic success or research conclusions, a validated protocol is not a recommendation but a necessity. This guide provides a technical framework for the validation of preservation protocols, focusing on the quantitative assessment of outcomes against the backdrop of known freezing-induced injuries.
Foundational Cell Injury Mechanisms During Freezing
A thorough understanding of freezing-induced cell injury is fundamental to developing and validating effective preservation protocols. The primary mechanisms of damage are mechanical and osmotic in nature, directly impacting cell viability and function.
- Intracellular Ice Formation (IIF): At rapid cooling rates, water within the cell does not have sufficient time to efflux and instead freezes internally. This IIF is almost universally lethal, causing mechanical damage to intracellular organelles and the plasma membrane. The osmotic rupture hypothesis suggests that the osmotic efflux of water during freezing can itself generate sufficient pressure to rupture the plasma membrane, creating pores that allow extracellular ice to propagate into the cytoplasm [113].
- Solution Effects Injury: At slow cooling rates, ice formation occurs predominantly in the extracellular space. This concentrates the solutes in the unfrozen fraction, creating a hypertonic environment that leads to severe cellular dehydration and volumetric shrinkage. This prolonged exposure to high solute concentrations and osmotic stress can damage membrane lipids and proteins [121].
- Eutectic Crystallization: Beyond electrolyte concentration, a further injury mechanism occurs upon eutectic crystallization, where certain salts in the solution simultaneously crystallize. Studies have shown a significant drop in post-thaw viability upon the occurrence of eutectic crystallization, suggesting direct mechanical damage to the cell membrane or intracellular eutectic formation [122].
Table 1: Primary Mechanisms of Freezing-Induced Cell Injury
| Mechanism | Cooling Rate | Primary Cause of Injury | Key References |
|---|---|---|---|
| Intracellular Ice Formation (IIF) | Rapid | Mechanical damage from internal ice crystals; Osmotic rupture of the plasma membrane. | [113] |
| Solution Effects Injury | Slow | Osmotic dehydration; Toxic solute concentration; Volumetric shrinkage. | [121] |
| Eutectic Crystallization | Very Slow/Specific Temperatures | Mechanical damage from salt crystallization; Intracellular eutectic formation. | [122] |
Major Preservation Methodologies and Their Validation
Different preservation strategies mitigate the aforementioned injury mechanisms through controlled physical and chemical interventions. The validation parameters must be tailored to the specific method and its intended application.
Cryopreservation
Cryopreservation involves cooling cells to sub-zero temperatures, typically below -100°C, to suspend all biological activity. The core principle is to navigate the cell through the temperature zone of maximum ice formation with minimal damage.
- Slow Freezing: This controlled-rate freezing method allows for controlled cellular dehydration. As extracellular ice forms, the cell loses water to the external hypertonic solution, thus minimizing the chance of IIF. It is the most widely adopted method, representing 67% of the cell freezing media demand in 2025 [123]. Validation requires optimization and strict control of the cooling rate (often 1°C/min) for different cell types.
- Vitrification: This is an "ice-free" preservation technique where high concentrations of cryoprotectants and ultra-rapid cooling are used to solidify the solution into a glassy, amorphous state. While it avoids ice crystallization, the challenges include cryoprotectant toxicity and the difficulty of scaling up for larger tissues [121]. Validation must confirm the absence of both ice formation and significant toxicity.
Static Cold Storage (SCS)
SCS involves storing biological materials at hypothermic temperatures (typically 0-4°C) in a specialized preservation solution. It is the cornerstone of short-term graft preservation. The method slows cellular metabolism but does not stop it, leading to risks over time, including cell edema, acidosis, and the production of reactive oxygen species (ROS) [121]. Validation for SCS is time-sensitive, focusing on maintaining viability and function within a defined, short-term window (e.g., hours to a few days).
Advanced Physical and Chemical Approaches
- Machine Perfusion: A dynamic alternative to SCS, machine perfusion provides continuous or intermittent circulation of an oxygenated preservation solution. This provides nutrients and removes waste, extending viable preservation times. Normothermic Machine Perfusion (NMP) maintains the graft at physiological temperatures (35-38°C), while Hypothermic Machine Perfusion (HMP) operates below 10°C [121]. Validation requires monitoring metabolic parameters and vascular function throughout the perfusion period.
- Chemical Fixation: Using cross-linking agents like glutaraldehyde preserves tissue structure for the long term but at the cost of cellular viability and regenerative capacity. It is prone to tissue deterioration, such as calcification, over time [121]. Validation here focuses on structural integrity and the reduction of antigenicity rather than cell viability.
- Decellularization: This process removes cellular content, leaving behind the extracellular matrix (ECM) scaffold, which is then often freeze-dried. This minimizes immune rejection and provides a platform for regenerative medicine [121]. Validation assesses the completeness of cell removal, the integrity of the ECM, and the biomechanical properties of the scaffold.
Table 2: Comparison of Major Preservation Methodologies
| Method | Temperature Range | Primary Application | Key Validation Metrics | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Slow Freezing | -150°C to -196°C | Stem cells, primary cells, gametes [124] | Post-thaw viability, recovery rate, apoptosis, functionality | Well-established, scalable, suitable for many cell types | Risk of solution effects injury, requires controlled-rate equipment |
| Vitrification | -150°C to -196°C | Oocytes, embryos, complex tissues [121] | Glass formation (ice-free state), structural integrity, function | Avoids ice crystal injury, high survival for sensitive cells | Cryoprotectant toxicity, technical complexity, limited sample volume |
| Static Cold Storage (SCS) | 0°C to 4°C | Solid organs, tissues for short-term storage [121] | ATP levels, membrane integrity, histology post-storage | Simple, low-cost, portable | Short storage window, ischemia-reperfusion injury |
| Machine Perfusion | 4°C (HMP) to 38°C (NMP) | Solid organs (heart, kidney, liver) [121] | Lactate metabolism, pH, vascular resistance, oxygen consumption | Extends preservation time, allows for viability assessment | Complex, expensive, requires specialized equipment |
| Decellularization/Freeze-dry | Room temp (post-process) | Tissue grafts (heart valves, vessels) [121] | DNA removal, ECM composition, mechanical strength, biocompatibility | Off-the-shelf availability, low immunogenicity | No living cells, potential for ECM damage during processing |
Core Validation Protocol Framework
A robust validation framework must systematically assess the protocol's performance against standardized benchmarks. The following workflow outlines the key stages from pre-preservation processing to final quality control.
Diagram 1: Validation Workflow
Experimental Methodologies for Key Assays
The following are detailed protocols for critical experiments cited in validation workflows.
- Protocol for Cryopreservation and Biobanking of Patient-Derived Cells: Adapted from recent peer-reviewed methodologies [125], this protocol involves suspending cells in a freezing medium, such as one containing 10% DMSO, and transferring them to cryovials. The vials are placed in an isopropanol freezing jar or a controlled-rate freezer to achieve a cooling rate of approximately -1°C/min until reaching -80°C, before long-term storage in liquid nitrogen vapor phase [-150°C or below] [126]. For thawing, vials are rapidly warmed in a 37°C water bath and the cell suspension is immediately diluted with pre-warmed culture medium to minimize cryoprotectant toxicity.
- Experimental Volume Measurements During Freezing: To directly observe osmotic responses, cryomicroscopy is employed. Cells are placed on a temperature-controlled stage and subjected to a defined freezing ramp. Time-lapse imaging captures real-time changes in cell volume as water effluxes during extracellular ice formation. This allows for the calculation of key membrane permeability parameters (Lp and Ps) [127].
- Assessing Osmotic Stress on Cell Cycle Dynamics: To investigate the effect of osmotic pressure beyond freezing, cells (e.g., FUCCI2 reporter lines) can be exposed to hyperosmotic media using solutes like PEG or sorbitol. Quantitative, time-lapse imaging then tracks the duration of cell cycle phases (G1, S/G2/M) and the emergence of growth-arrested subpopulations under mild (380 mOsm kg⁻¹) and high (460 mOsm kg⁻¹) stress compared to an isotonic control (320 mOsm kg⁻¹) [128].
Quantitative Metrics and Acceptance Criteria
Validation requires moving beyond simple viability to a multi-parameter assessment. The following table outlines key quantitative metrics.
Table 3: Key Validation Metrics and Target Acceptance Criteria
| Validation Category | Specific Metric | Target Acceptance Criteria (Example) | Measurement Technique |
|---|---|---|---|
| Viability & Yield | Post-thaw viability | >80% for stem cells; >70% for primary cells | Flow cytometry (Annexin V/PI), Trypan Blue exclusion |
| Total cell recovery | >75% of pre-freeze count | Automated cell counter | |
| Phenotype & Function | Surface marker expression | >85% retention of key markers (e.g., CD34+, CD45-) | Flow cytometry |
| Metabolic activity | >80% of unfrozen control after 72h culture | MTT/XTT assay, ATP assay | |
| Differentiation potential | Retention of multi-lineage capacity | Directed differentiation assays | |
| Cell cycle dynamics | Return to normal distribution within 48h | FUCCI2 imaging, PI staining [128] | |
| Molecular Integrity | Genomic integrity | No significant increase in DNA damage vs. control | Comet assay, Karyotyping |
| Transcriptomic profile | High correlation (R² > 0.9) with unfrozen control | RNA-Seq, Microarray | |
| Proteomic profile | Preservation of key protein expression and modification | Western blot, Mass spectrometry |
The Scientist's Toolkit: Research Reagent Solutions
A successful validation pipeline relies on a suite of reliable reagents and equipment. The selection below is based on current market trends and widespread adoption.
Table 4: Essential Research Reagents and Equipment for Preservation Validation
| Item | Function | Key Considerations & Examples |
|---|---|---|
| Cryopreservation Media | Provides a protective environment during freezing; contains buffers, proteins, and cryoprotectants. | DMSO is the gold standard, used in ~70.9% of media [123]. Serum-free and xeno-free formulations are critical for clinical applications. |
| Programmable Freezer | Enables controlled-rate freezing, critical for optimizing cooling rates and minimizing ice crystal injury. | Equipment segment dominates the market [124]. New large-capacity models support scale-up for cell therapy production. |
| Liquid Nitrogen Storage | Provides ultra-low temperature (-196°C) for long-term storage, halting all biological activity. | Vapor phase storage (46% market share) is preferred to minimize contamination risk vs. liquid phase [129]. |
| Viability/Cytotoxicity Kits | To accurately assess post-thaw cell health and distinguish live, apoptotic, and necrotic populations. | Annexin V/Propidium Iodide (PI) kits are standard. Alternative dyes include 7-AAD and Calcein AM. |
| Cell Cycle Reporter System | For real-time, non-invasive tracking of cell cycle progression and arrest in response to stress. | FUCCI2 (Fluorescent Ubiquitination-based Cell Cycle Indicator) enables single-cell analysis [128]. |
| Osmolality Meter | To precisely measure and adjust the osmolarity of preservation solutions, a critical parameter. | Essential for creating hyperosmotic stress models and ensuring consistency of freezing and culture media. |
The validation of preservation protocols is a complex, multi-faceted process that sits at the intersection of cell biology, biophysics, and regulatory science. It requires a deep understanding of the mechanical and osmotic injuries inflicted during freezing and a systematic approach to mitigating them. By adopting the structured framework outlined in this guide—from foundational science and method selection to rigorous experimental protocols and multi-parameter quality control—researchers and drug development professionals can ensure that their preserved biological samples are not merely "viable" but truly retain the functional and molecular characteristics required for robust clinical applications and groundbreaking research. As the field advances with innovations like nano-warming, advanced cryoprotectants, and AI-integrated storage monitoring [121] [129], validation protocols must similarly evolve to guarantee that the immense potential of cell-based therapies is fully realized.
Quality Control Metrics for Cell Therapy Products and Biopharmaceuticals
The cryopreservation of cell therapy products and biopharmaceuticals introduces significant mechanical and osmotic stresses that directly impact critical quality attributes. During freezing, water undergoes a phase transition to ice, creating a dual injury paradigm: mechanical damage from ice crystal formation and osmotic stress from solute concentration effects [3] [80]. These cryoinjuries can compromise membrane integrity, disrupt intracellular architecture, and alter cellular function, ultimately threatening product safety and efficacy [62]. Understanding these fundamental mechanisms is essential for developing robust quality control metrics that accurately predict post-thaw product performance.
The cellular response to freezing begins when temperatures drop below 0°C. Extracellular ice formation excludes solutes, creating a hypertonic environment that draws water out of cells through osmosis [3]. This dehydration causes cell shrinkage and increases intracellular solute concentrations to potentially toxic levels—a phenomenon known as "solution effects" injury [80]. If cooling occurs too rapidly, water cannot exit cells quickly enough, leading to lethal intracellular ice formation (IIF) that mechanically disrupts organelles and membranes [80] [62]. The balance between these two damage mechanisms follows a reverse U-shaped curve, where optimal cooling rates minimize both excessive dehydration and IIF [62].
For cell-based therapeutics, these cryoinjuries manifest not only as reduced viability but potentially more critically as altered functionality. Research demonstrates that natural killer (NK) cells show reduced cytotoxic activity after thawing, while mesenchymal stem cells (MSCs) exhibit increased apoptosis levels [63]. These functional impairments often remain undetected in standard viability tests, creating a dangerous discrepancy between measured viability and therapeutic potential [63]. Therefore, quality control strategies must extend beyond traditional membrane integrity assessments to include functional and potency assays that reflect the product's intended biological activity.
Key Quality Attributes and Their Measurement
Critical Quality Attributes (CQAs) for Cryopreserved Products
Table 1: Essential Quality Attributes for Cryopreserved Cell Therapies
| Quality Attribute | Significance in Cryopreservation | Standard Assessment Methods | Technological Advances |
|---|---|---|---|
| Viability | Measures membrane integrity compromised by ice crystals and osmotic stress [80] | Trypan blue exclusion, flow cytometry with viability dyes [80] | In situ dielectric spectroscopy for real-time monitoring [130] |
| Potency | Indicates biological function retention post-thaw; often diminished despite adequate viability [63] | Cell-specific functional assays (e.g., cytotoxic activity, differentiation potential) [131] | AI-based analytics for predicting functional potency [130] |
| Sterility | Ensures absence of microbial contamination introduced during processing [131] | Sterility testing per pharmacopeia standards [131] | Rapid microbiological methods with reduced turnaround time [130] |
| Identity/Purity | Verifies cell population consistency after freezing-induced selective pressures [63] | Flow cytometry, PCR, morphological analysis [132] | Automated liquid handling robots for consistent analysis [130] |
| Apoptosis/Necrosis | Quantifies freezing-induced programmed cell death versus necrotic death [63] | Annexin V/Propidium Iodide staining [132] | Multi-parameter flow cytometry with AI-based gating [130] |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Materials for Cryopreservation Quality Assessment
| Reagent/Material | Function in QC Testing | Application Notes |
|---|---|---|
| Cryoprotective Agents (CPAs) | Protect against ice crystal damage and osmotic stress [3] | DMSO toxicity varies by batch; requires qualification [132] |
| Viability Assay Kits | Distinguish live/dead cells based on membrane integrity [80] | Trypan blue misses early apoptosis; combine with functional assays [63] |
| Flow Cytometry Reagents | Characterize cell identity, purity, and apoptosis [132] | AI-based gating tools (e.g., BD ElastiGate) improve reproducibility [130] |
| Sterility Testing Kits | Detect microbial contamination in final product [131] | Rapid microbiological methods reduce turnaround time [130] |
| Host Cell Protein Assays | Detect residual process-related impurities [133] | Activity-based protein profiling identifies enzymatically active HCPs [133] |
| Automated Liquid Handlers | Standardize sample preparation for QC testing [130] | Reduce variability in labor-intensive analytical steps [130] |
Analytical Methods for Assessing Cryopreservation Outcomes
Structural and Functional Assessment Techniques
The post-thaw assessment of cell therapies requires a multi-parametric approach that evaluates both structural integrity and biological function. Conventional membrane integrity tests using dye exclusion methods (e.g., trypan blue) provide a basic viability percentage but fail to detect more subtle cryoinjuries such as cytoskeletal disorganization or organelle dysfunction [80] [63]. Multi-parameter flow cytometry offers enhanced capability by simultaneously measuring membrane integrity, apoptosis markers (Annexin V), and cell surface markers for identity and purity [132]. Emerging technologies like in situ dielectric spectroscopy enable real-time, non-invasive monitoring of cell density and viability during processing, potentially revolutionizing current QC practices by providing continuous process data rather than single timepoint measurements [130].
Functional potency assays are particularly critical for cell therapies, as they represent the product's intended biological effect. For immunotherapies such as CAR-T cells, this typically involves target cell killing assays that quantify cytotoxic potential [131]. For stem cell products, differentiation assays measuring the ability to form specific lineages provide crucial potency data [132]. These functional assessments are especially relevant in the context of cryopreservation, as freezing and thawing can disproportionately impact functional pathways while leaving basic viability intact. Research demonstrates that mesenchymal stem cells may maintain membrane integrity post-thaw while showing significantly reduced engraftment potential and increased apoptosis—a concerning discrepancy for therapeutic efficacy [63].
Emerging Analytical Technologies
The field of quality control is rapidly evolving with the integration of advanced analytical technologies that provide deeper characterization of cryopreservation impacts. Process Analytical Technology (PAT) approaches enable real-time monitoring of critical process parameters, facilitating better control of the freezing and thawing processes that directly influence product quality [133]. The Multi-Attribute Method (MAM) using liquid chromatography-mass spectrometry (LC-MS) represents a significant advancement for simultaneously monitoring multiple product quality attributes, including post-translational modifications that may be affected by freezing stresses [133].
Artificial intelligence and machine learning are increasingly applied to quality control challenges. AI-based gating tools for flow cytometry analysis standardize data interpretation across operators and sites, reducing a significant source of variability in cell product characterization [130]. Digital twin technology creates virtual replicas of cryopreservation processes, enabling simulation and optimization of parameters before implementation in manufacturing [130]. These digital approaches are particularly valuable for autologous cell therapies with limited batch sizes for process development.
Experimental Protocols for Evaluating Cryopreservation Effects
Protocol: Comprehensive Post-Thaw Cell Quality Assessment
This protocol outlines a standardized methodology for evaluating the impact of cryopreservation on cell therapy products, specifically addressing both mechanical and osmotic injury mechanisms.
Materials and Equipment:
- Pre-cryopreservation cell sample (reference control)
- Cryopreserved cell sample
- Complete culture medium
- Cryoprotectant solution (e.g., 10% DMSO)
- Water bath (37°C)
- Centrifuge
- Flow cytometer with viability dyes (e.g., PI, 7-AAD) and Annexin V
- Hemocytometer or automated cell counter
- Functional assay reagents (cell-type specific)
Procedure:
- Thawing and Preparation:
- Rapidly thaw cryopreserved cells in a 37°C water bath with gentle agitation until only a small ice crystal remains (approximately 2-3 minutes) [80].
- Transfer cells to a pre-labeled tube and slowly dilute 1:10 with pre-warmed culture medium to minimize osmotic shock during cryoprotectant removal [132].
- Centrifuge at 300-400 × g for 5-7 minutes and carefully aspirate supernatant containing cryoprotectant.
- Resuspend cell pellet in appropriate volume of culture medium.
Viability and Recovery Assessment:
- Remove an aliquot of cells for counting and viability assessment.
- Mix cells with trypan blue (1:1 ratio) and count using a hemocytometer or automated cell counter.
- Calculate total viable cell recovery: (Post-thaw viable cell count / Pre-freeze viable cell count) × 100 [132].
- Note morphological changes under microscopy, including cell swelling, membrane blebbing, or fragmentation.
Flow Cytometric Analysis:
- Prepare aliquots of 1 × 10^6 cells for multi-parameter flow cytometry.
- Stain cells according to manufacturer protocols for:
- Viability dye (e.g., PI or 7-AAD)
- Annexin V for apoptosis detection
- Cell-specific identity markers (e.g., CD markers for immune cells)
- Include appropriate compensation controls and unstained samples.
- Analyze using flow cytometry, collecting at least 10,000 events per sample.
Functional Potency Assessment:
- Perform cell-specific functional assays based on therapeutic mechanism:
- For immune effector cells: Co-culture with target cells at various effector:target ratios and measure target cell killing after 4-24 hours [131].
- For stem cells: Assess differentiation potential by culturing in lineage-specific media and evaluating marker expression after 7-14 days.
- For secretory cells: Measure specific protein production (e.g., cytokines, hormones) under stimulated conditions using ELISA or similar methods.
- Compare functional potency of post-thaw cells to pre-freeze controls.
- Perform cell-specific functional assays based on therapeutic mechanism:
Data Analysis and Interpretation:
- Calculate percentage values for viability, apoptosis, and identity markers.
- Compare post-thaw recovery and function to pre-freeze reference samples.
- Note any subpopulation shifts that may indicate selective loss of specific cell types during cryopreservation.
Protocol: Osmotic Stress Evaluation During Cryoprotectant Exposure
This protocol specifically assesses cellular response to osmotic stress during cryoprotectant addition and removal, key steps in the cryopreservation process.
Materials and Equipment:
- Cell sample
- Cryoprotectant solutions (e.g., DMSO, glycerol, or commercial alternatives)
- Isotonic buffer
- Microscope with imaging capability
- Cell counting equipment
Procedure:
- Cell Preparation:
- Prepare a single-cell suspension at a concentration of 1 × 10^6 cells/mL in isotonic buffer.
- Divide into aliquots for different cryoprotectant conditions.
Cryoprotectant Exposure:
- Gradually add cryoprotectant solutions to cell suspensions in a stepwise manner (e.g., 2.5% increments every 2-3 minutes) to minimize osmotic shock [132].
- After each addition, observe cell morphology changes under microscopy and record images.
- Incubate cells with final cryoprotectant concentration (typically 10% for DMSO) for 15 minutes at 4°C to simulate standard cryopreservation conditions.
Cryoprotectant Removal:
- Gradually dilute samples with isotonic buffer in a stepwise manner.
- Observe and document morphological changes during dilution.
Assessment:
- Count cells before and after the procedure to determine recovery rate.
- Assess viability using trypan blue exclusion or similar method.
- Document volumetric changes and morphological alterations throughout the process.
Process Optimization Strategies
Controlling Freezing and Thawing Parameters
Optimizing the thermal parameters of cryopreservation is essential for minimizing freezing-induced cellular damage. The cooling rate represents a critical process parameter that directly influences the balance between osmotic dehydration and intracellular ice formation. For many mammalian cell types, cooling rates of approximately 1°C per minute provide optimal recovery by allowing sufficient water efflux to avoid lethal intracellular ice formation while limiting excessive dehydration [3] [80]. However, cell-specific optimization is necessary, as different cell types demonstrate varying membrane permeability and tolerance to osmotic stress. Research indicates that rapid cooling is associated with better outcomes for oocytes, pancreatic islets, and embryonic stem cells, while slow cooling is recommended for hepatocytes, hematopoietic stem cells, and mesenchymal stem cells [3].
The thawing process requires equally careful control to minimize damaging ice recrystallization. Warming rates of 60-80°C per minute are typically recommended to rapidly transition through the dangerous temperature zone (-15°C to 0°C) where ice crystal growth occurs most readily [80]. Controlled-rate thawing devices provide more consistent results compared to uncontrolled methods like room temperature placement. The temperature history during storage and transport also significantly impacts product quality. Transient warming events above the glass transition temperature of water (approximately -135°C) can trigger microscopic melting and recrystallization, leading to progressive damage to cellular structures [80]. Implementing automated storage systems with robotic retrieval can minimize these temperature fluctuations by reducing human intervention during sample handling.
Advanced Cryopreservation Strategies
Emerging cryopreservation strategies aim to better control ice formation and mitigate cryoprotectant toxicity. Vitrification approaches using high CPA concentrations and ultra-rapid cooling completely avoid ice formation by transitioning cells directly into a glassy state [62]. While effective for stress-sensitive cells like oocytes and stem cells, conventional vitrification requires high CPA concentrations (6-8 M) that introduce significant toxicity concerns [62]. Low-CPA vitrification strategies address this limitation by combining increased cooling rates with reduced CPA concentrations, though this approach requires specialized equipment to achieve the necessary thermal transfer rates [62].
Encapsulation technologies represent another promising advancement, where cells are immobilized within protective hydrogel matrices such as alginate or polyethylene glycol (PEG) [134]. These biomaterials provide a physical barrier against ice crystal penetration and help regulate osmotic balance by controlling water and cryoprotectant diffusion. Studies demonstrate that human mesenchymal stem cells encapsulated in alginate hydrogels showed 92% post-thaw viability compared to 67% for non-encapsulated controls [134]. Microfluidic encapsulation techniques further enhance this approach by producing highly uniform hydrogel beads with precise control over size and composition, leading to more consistent freezing and thawing outcomes across cell populations [134].
Workflow Visualization: Quality Control Pathway for Cryopreserved Cell Therapies
The diagram below illustrates the integrated quality control pathway for cryopreserved cell therapies, highlighting critical assessment points and decision gates throughout the process.
Quality Control Pathway for Cryopreserved Cell Therapies
This workflow emphasizes the critical assessment points where mechanical and osmotic stresses from cryopreservation are evaluated. The parallel assessment of structural integrity, functional potency, and identity/purity ensures comprehensive evaluation of freezing-induced damage, with each category requiring satisfactory results for product release.
The expanding cell and gene therapy market, projected to grow at a CAGR of 25.74% from 2025 to 2034 [131], underscores the critical need for robust quality control frameworks specifically designed to address cryopreservation-induced stresses. Traditional quality metrics focusing primarily on viability provide insufficient assurance of therapeutic efficacy, as significant functional impairments can persist in cells that maintain membrane integrity [63]. A comprehensive quality assessment strategy must therefore integrate multiple orthogonal methods that collectively evaluate structural integrity, functional potency, and population purity.
Emerging technologies including artificial intelligence, process analytical technology (PAT), and advanced mass spectrometry methods are transforming quality control paradigms for cryopreserved products [133] [130]. These approaches enable real-time monitoring of critical quality attributes during the freezing process itself, facilitating proactive quality management rather than retrospective assessment. The development of digital twin technology for simulating cryopreservation processes represents a particularly promising approach for optimizing parameters without consuming valuable cellular material [130]. As the field advances, the integration of these sophisticated analytical methods with improved understanding of fundamental cryoinjury mechanisms will enable more predictive quality models that ensure the consistent production of safe and effective cryopreserved cell therapies.
Conclusion
The successful preservation of cellular integrity during freezing requires a nuanced understanding of the interconnected mechanical and osmotic stresses that cells endure. By integrating foundational knowledge of biophysical damage mechanisms with advanced methodological approaches and rigorous validation, researchers can develop optimized preservation protocols that maximize cell survival and functionality. Future directions in cryobiology will likely focus on cell-type specific preservation strategies, the development of less toxic cryoprotectant solutions, and advanced technologies that precisely control ice formation and mitigate mechanical stress. These advancements hold significant promise for enhancing the efficacy of cell-based therapies, biobanking, and pharmaceutical development by ensuring the reliable preservation of cellular function across diverse biomedical applications.
References
- 1. Quantitative assessment of the impact of cryopreservation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7734731/]
- 2. Freezing of living cells: mechanisms and implications [https://pubmed.ncbi.nlm.nih.gov/6383068/]
- 3. Cryopreservation: An Overview of Principles and Cell- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7995302/]
- 4. Probing mechanisms of cryopreservation damage to ... [https://pubmed.ncbi.nlm.nih.gov/39918490/]
- 5. Improving Cell Recovery: Freezing and Thawing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8909336/]
- 6. Impact of Freezing and Freeze Drying on Lactobacillus ... [https://www.mdpi.com/2304-8158/14/10/1817]
- 7. Cell Freezing Protocols | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/cell-culture-protocols/freezing-cells.html]
- 8. Membrane-targeted DNA frameworks with biodegradability ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925003130]
- 9. Experimental Cryosurgery Investigations In Vivo - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2821701/]
- 10. Mechanisms of intracellular ice formation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1280746/]
- 11. Chemical approaches to cryopreservation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9294745/]
- 12. Microscopic Mechanism and Kinetics of Ice Formation at ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4939469/]
- 13. Modeling and typical cases analyze at the cell-scale of ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224025000161]
- 14. Kinetics and activation energy of recrystallization ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224008000229]
- 15. Tricine as a Novel Cryoprotectant with Osmotic Regulation ... [https://www.mdpi.com/1422-0067/23/15/8462]
- 16. The need for novel cryoprotectants and cryopreservation ... [https://www.sciencedirect.com/science/article/abs/pii/S0304416520302609]
- 17. Role of Cells in Freezing-Induced Cell-Fluid-Matrix ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3708713/]
- 18. Cryopreservation of tissues by slow-freezing using an ... [https://www.nature.com/articles/s41598-022-23913-3]
- 19. Osmotic properties of T cells determined by flow imaging ... [https://www.sciencedirect.com/science/article/abs/pii/S001122402300086X]
- 20. Effect of freezing on cell viability and mechanical strength ... [https://experts.umn.edu/en/publications/effect-of-freezing-on-cell-viability-and-mechanical-strength-of-b/]
- 21. Osmotic Dehydration, Drying Kinetics, and Quality ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8834474/]
- 22. Freezing induced incorporation of betaine in lipid ... [https://www.nature.com/articles/s41467-025-60040-9]
- 23. Freezing negatively impacts the interlamellar mechanical ... [https://www.sciencedirect.com/science/article/pii/S002192902500291X]
- 24. Osmotic pressure‐induced calcium response states - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12485884/]
- 25. Extracellular freezing-induced mechanical stress and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2652538/]
- 26. Evaluation of Cell Lysis Due to Ice Crystals in Cell Freezing [https://biomedres.us/fulltexts/BJSTR.MS.ID.006716.php]
- 27. New insights into the mechanism of freeze-induced ... [https://www.sciencedirect.com/science/article/abs/pii/S0963996922008158]
- 28. The effects of osmotic stress on the structure and function of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3616882/]
- 29. Dynamic response of the cell traction force to osmotic shock [https://www.nature.com/articles/s41378-023-00603-2]
- 30. Lamellar-to-hexagonalII phase transitions in the plasma ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC391926/]
- 31. Effects of freezing on membranes and proteins in LNCaP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1994664/]
- 32. The mechanism of lamellar-to-inverted hexagonal phase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1275708/]
- 33. The lamellar to hexagonal II (HII) phase transition [https://link.springer.com/article/10.1007/BF01869487]
- 34. Mode of action of the COR15a gene on the freezing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC24414/]
- 35. Membrane hydration correlates to cellular biophysics ... [https://www.sciencedirect.com/science/article/pii/S0005273609000418]
- 36. Cell surface area regulation and membrane tension [https://pubmed.ncbi.nlm.nih.gov/11220366/]
- 37. Surface area regulation: underexplored yet crucial in cell ... [https://www.nature.com/articles/nrm2419-c2]
- 38. Regulation of the Total Cell Surface Area in Dividing ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2020.00238/full]
- 39. Freezing tolerance and tolerance to de-acclimation of ... [https://www.nature.com/articles/s41598-023-47318-y]
- 40. Different storage and freezing protocols for extracellular vesicles [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-024-04005-7]
- 41. Cryopreservation: A Review Article - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9756256/]
- 42. Cellular cryobiology: thermodynamic and mechanical effects [https://www.sciencedirect.com/science/article/abs/pii/S014070070000027X]
- 43. Cryopreservation as a Key Element in the Successful ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2020.592242/full]
- 44. Effects, methods and limits of the cryopreservation on ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-024-03954-3]
- 45. Slow freezing [https://bio-protocol.org/exchange/minidetail?id=19079401&type=30]
- 46. Establishment of a controlled slow freezing-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8978083/]
- 47. Optimization of freezing and thawing protocols for human ... [https://www.sciencedirect.com/science/article/pii/S0011224025000513]
- 48. What's the difference between slow freezing and vitrification? [https://extendfertility.com/slow-freezing-vs-vitrification/]
- 49. Vitrification versus slow freezing gives excellent survival ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2729856/]
- 50. Efficacy and safety of a one-step warming protocol ... [https://www.medrxiv.org/content/10.1101/2025.10.02.25337068v1.full-text]
- 51. The role of heat shock protein 70 on oocyte apoptosis during ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12613207/]
- 52. Cryoprotectant Toxicity: Facts, Issues, and Questions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4620521/]
- 53. Natural Cryoprotective and Cytoprotective Agents in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9145333/]
- 54. Mechanisms of freezing damage [https://pubmed.ncbi.nlm.nih.gov/3332492/]
- 55. Cell freezing and the biology of inexorability - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11564080/]
- 56. Mechanisms of stabilization, damage during freezing [https://biocor.umn.edu/research/preservation_mechanisms]
- 57. 5 Key Differences Between Cryoprotectant Types [https://allanchem.com/key-differences-cryoprotectant-types/]
- 58. Cryoprotectant - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/cryoprotectant]
- 59. Chemical approaches to cryopreservation [https://www.nature.com/articles/s41570-022-00407-4]
- 60. Cryoprotective effects of grape seed protein hydrolysates in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12359270/]
- 61. Tissue Cryoprotectant 2025-2033 Overview [https://www.archivemarketresearch.com/reports/tissue-cryoprotectant-328197]
- 62. Advanced technologies for the preservation of mammalian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8765766/]
- 63. From Freezer to Failure? The Hidden Risks ... [https://cellbox-solutions.com/cellbox-blog/from-freezer-to-failure-the-hidden-risks-of-cryopreservation-in-cell-therapy]
- 64. The impact of initial cooling rates on cell preservation in frozen ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12408769/]
- 65. Interrupted cooling protocols in cryopreservation: A review ... [https://www.sciencedirect.com/science/article/abs/pii/S0011224025001452]
- 66. Cryopreservation - Key Survey Findings from the ISCT ... [https://www.isctglobal.org/telegrafthub/blogs/ken-ip1/2025/05/28/cryopreservation-key-survey-findings-from-the-isct]
- 67. Slow Freezing Coupled Static Magnetic Field Exposure ... - NCBI [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3592815/]
- 68. Impact of controlled ice nucleation on intracellular ... [https://pubmed.ncbi.nlm.nih.gov/39265855/]
- 69. Controlled nucleation in freeze-drying [https://www.europeanpharmaceuticalreview.com/article/15427/controlled-nucleation-in-freeze-drying/]
- 70. Magnetic Field (MF) Assisted Freezing in Food Preservation [https://pubmed.ncbi.nlm.nih.gov/40556131/]
- 71. Controlled nucleation of crystallization process as an ... [https://www.sciencedirect.com/science/article/abs/pii/S0032591022002285]
- 72. Combined vacuum osmotic dehydration by pomegranate ... [https://www.nature.com/articles/s41598-025-19708-x]
- 73. Osmotic stress as a mechanism of freezing injury [https://www.sciencedirect.com/science/article/pii/001122407190040X]
- 74. Controlled freeze drying, storage and enteric coated ... [https://www.sciencedirect.com/science/article/pii/S2405844024044384]
- 75. Protocols and Best Practices for Freezing Cells [https://www.stemcell.com/cryopreservation-basics-protocols-and-best-practices-for-freezing-cells]
- 76. Examining the Effect of Freezing Temperatures on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10972471/]
- 77. ENHANCED BACTERIAL VIABILITY OF FREEZE-DRIED ... [https://www.nal.usda.gov/research-tools/food-safety-research-projects/enhanced-bacterial-viability-freeze-dried-probiotics]
- 78. Effect of freezing rate on water state in strawberry [https://www.sciencedirect.com/science/article/abs/pii/S0308814625036593]
- 79. Effects of Freezing, Frozen Storage and Thawing on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12331523/]
- 80. Protecting Cell Viability During Cryopreservation [https://www.azenta.com/learning-center/blog/considerations-protecting-cell-viability-during-cryopreservation]
- 81. Optimizing cryopreservation strategies for scalable cell therapies [https://pmc.ncbi.nlm.nih.gov/articles/PMC11659802/]
- 82. Insights on the role of cryoprotectants in enhancing the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11729100/]
- 83. Cryoprotectant Toxicity: Balancing Safety and Efficacy [https://allanchem.com/cryoprotectant-toxicity-safety-efficacy/]
- 84. Efficacy assessment of different cryoprotectants for ... [https://www.nature.com/articles/s41598-024-71529-6]
- 85. Cryoprotectant [https://en.wikipedia.org/wiki/Cryoprotectant]
- 86. Model-Guided Design and Optimization of CPA Perfusion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10518287/]
- 87. Impact of Freezing and Freeze Drying on Lactobacillus ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12111118/]
- 88. A laboratory-friendly protocol for freeze-drying sample ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1523712/full]
- 89. Lipid Properties and Metabolism in Response to Cold [https://pubmed.ncbi.nlm.nih.gov/40195263/]
- 90. Physiological and Molecular Mechanisms of Freezing in ... [https://www.sciencedirect.com/org/science/article/pii/S0031945725001005]
- 91. Cold adaptation strategies in plants—An emerging role of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9459425/]
- 92. Quantitative imaging of lipid transport in mammalian cells [https://www.nature.com/articles/s41586-025-09432-x]
- 93. Quantitative retention of membrane lipids in the freeze- ... [https://pubmed.ncbi.nlm.nih.gov/17876599/]
- 94. Quantitative retention of membrane lipids in the freeze- ... [https://link.springer.com/article/10.1007/s00418-007-0341-3]
- 95. Parallel Effects of Freezing and Osmotic Stress on the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1062102/]
- 96. Brief Overview of Ice Nucleation - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7828621/]
- 97. Effect of diffusion kinetics on the ice nucleation temperature ... [https://www.nature.com/articles/s41598-022-20797-1]
- 98. The Silent Threat in Cell Therapy Development: Transient ... [https://www.biolifesolutions.com/resource-center/insights-blog/the-silent-threat-in-cell-therapy-development-transient-warming-events/]
- 99. Freezing-driven ionic charge imbalance leads to pore ... [https://www.sciencedirect.com/science/article/abs/pii/S0010482525003117]
- 100. Walking on thin ice: controlled freezing & thawing of pharmaceutical... [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/2677/walking-on-thin-ice-controlled-freezing-thawing-of-pharmaceutical-active-molecules]
- 101. Cell Culture – Why are my cells not attaching or proliferating after... [https://info.abmgood.com/cell-culture-thawing-protocol]
- 102. Freeze-drying of red blood cells: how useful are freeze/ thaw ... [https://pubmed.ncbi.nlm.nih.gov/10600256/]
- 103. Optimizing Sperm Cryopreservation from Four Endangered ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561652/]
- 104. Elamipretide enhances post-thaw rooster sperm quality by ... [https://www.nature.com/articles/s41598-025-09943-7]
- 105. Impact of pre- and post-thaw processing on recovery ... [https://pubmed.ncbi.nlm.nih.gov/41026081/]
- 106. 5-aminolevulinic acid improves drake post-thaw sperm ... [https://www.sciencedirect.com/science/article/pii/S0032579125012362]
- 107. Advancing Cancer Cell-Based Assays with Assay-Ready ... [https://www.thermofisher.com/blog/behindthebench/advancing-cancer-cell-based-assays-with-assay-ready-tumoroids/]
- 108. Cryopreservation and culture strategies for testicular tissue ... [https://www.frontiersin.org/journals/veterinary-science/articles/10.3389/fvets.2025.1638248/full]
- 109. Equivalent outcomes of human oocytes after vitrification or ... [https://pubmed.ncbi.nlm.nih.gov/40234976/]
- 110. Equivalent outcomes of human oocytes after vitrification or ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11998440/]
- 111. Comparison of vitrification and slow freezing for ... [https://www.msjonline.org/index.php/ijrms/article/view/1810]
- 112. Comparative effectiveness of vitrification and slow freezing ... [https://bmcwomenshealth.biomedcentral.com/articles/10.1186/s12905-024-03505-1]
- 113. The osmotic rupture hypothesis of intracellular freezing injury [https://pmc.ncbi.nlm.nih.gov/articles/PMC1275720/]
- 114. Raman cryomicroscopic imaging and sample holder ... [https://experts.umn.edu/en/publications/raman-cryomicroscopic-imaging-and-sample-holder-for-spectroscopic]
- 115. Time-deterministic cryo-optical microscopy | Light [https://www.nature.com/articles/s41377-025-01941-8]
- 116. Raman microscopy of cryofixed biological specimens for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11633761/]
- 117. Expanding the cryoprotectant toolbox in biomedicine by ... [https://www.sciencedirect.com/science/article/abs/pii/S073497502500031X]
- 118. Proteomic approach for evaluating cryoprotectant ... [https://www.nature.com/articles/s41598-025-00534-0]
- 119. Unraveling the Cryoprotective Molecular Mechanisms ... [https://pubmed.ncbi.nlm.nih.gov/40937661/]
- 120. The influence of magnetic field-assisted osmotic ... [https://www.sciencedirect.com/science/article/abs/pii/S0140700725004153]
- 121. Tissue Preservation and Access: Modern Innovation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12113189/]
- 122. Direct cell injury associated with eutectic crystallization during ... [https://pubmed.ncbi.nlm.nih.gov/14969678/]
- 123. Cell Freezing Media Market | Global Market Analysis Report [https://www.futuremarketinsights.com/reports/cell-freezing-media-market]
- 124. Cell Cryopreservation Market Size & Share Outlook to 2030 [https://www.mordorintelligence.com/industry-reports/cell-cryopreservation-market]
- 125. Protocol for the processing, cryopreservation, and ... [https://www.sciencedirect.com/science/article/pii/S2666166724004520]
- 126. Ultra-freezing and sample preservation in clinical biobanks [https://bexen.com/en/bioservices-en/ultra-freezing-biobank-sample-preservation/]
- 127. Osmotic response of individual cells during freezing [https://www.sciencedirect.com/science/article/abs/pii/0011224083900603]
- 128. Osmotic pressure modulates single cell cycle dynamics ... [https://www.nature.com/articles/s41598-021-92054-w]
- 129. Cryopreservation & Platelet Storage Technologies Market [https://www.globenewswire.com/news-release/2025/10/15/3167303/0/en/Cryopreservation-Platelet-Storage-Technologies-Market.html]
- 130. Keep An Eye On These Analytical And Monitoring Trends ... [https://www.bioprocessonline.com/doc/keep-an-eye-on-these-analytical-and-monitoring-trends-in-0001]
- 131. Cell & Gene Therapy Quality Control Market Grows at ... [https://www.towardshealthcare.com/insights/cell-and-gene-therapy-quality-control-market-sizing]
- 132. Impact of Freeze–Thaw Processes on the Quality of Cells [https://www.insights.bio/cell-and-gene-therapy-insights/journal/article/365/Impact-of-FreezeThaw-Processes-on-the-Quality-of-Cells]
- 133. Next-Generation Analytical Methods - 2025 Archive [https://www.bioprocessingeurope.com/25/analytical-methods]
- 134. Encapsulation for efficient cryopreservation [https://www.degruyterbrill.com/document/doi/10.1515/fzm-2025-0008/html?srsltid=AfmBOoohDuXNQPtDx8Fgfn0shZZy2e9mZ4dqYD2jyh5cyO2Lo8GDijX0]