Charge Variant Analysis in mAbs: A Comprehensive Guide from Characterization to Control
This article provides a comprehensive overview of charge variant analysis for monoclonal antibodies (mAbs), a critical requirement in biopharmaceutical development.
Charge Variant Analysis in mAbs: A Comprehensive Guide from Characterization to Control
Abstract
This article provides a comprehensive overview of charge variant analysis for monoclonal antibodies (mAbs), a critical requirement in biopharmaceutical development. It covers the foundational knowledge of charge heterogeneity, including the common post-translational modifications that generate acidic and basic variants. The content details state-of-the-art methodological approaches for separation and characterization, such as ion-exchange chromatography (IEC), capillary isoelectric focusing (ciIEF), and mass spectrometry. Furthermore, it discusses advanced troubleshooting, optimization strategies for controlling charge profiles during bioprocessing, and the critical step of validating the impact of charge variants on biological activity and stability to ensure product quality, safety, and efficacy for researchers and drug development professionals.
Understanding mAb Charge Heterogeneity: Origins, Impact, and Critical Quality Attributes
Recombinant monoclonal antibodies (mAbs) are not single, uniform entities but exist as heterogeneous mixtures of multiple structural variants. Charge heterogeneity is a common feature of therapeutic mAbs, where variants differ in their net surface charge or isoelectric point (pI) [1] [2]. These charge variants are typically categorized by their chromatographic or electrophoretic behavior relative to the predominant form of the antibody. Species with a lower intrinsic pI than the main species are termed acidic variants, while those with a higher pI are termed basic variants [2]. The well-characterized, predominant form is referred to as the main species [1]. Understanding the chemical nature and biological impact of these variants is a critical aspect of biopharmaceutical development, as they can influence the stability, efficacy, and safety of the therapeutic product [3] [4].
Definition and Characterization of Species
The separation of a mAb sample typically reveals three key regions: the acidic variants, the main species, and the basic variants. The following table summarizes their core definitions and common modifications.
Table 1: Core Definitions of Charge Variant Species
| Species | Definition | Common Modifications & Causes |
|---|---|---|
| Acidic Variants | Species with a lower isoelectric point (pI) than the main species [2]. | Deamidation, glycation, sialylated glycans, oxidation, fragmentation (low molecular weight species), trisulfide bonds, and succinimation [3] [2]. |
| Main Species | The well-understood, predominant form of the antibody [1]. | Typically lacks C-terminal lysine on heavy chains and is glycosylated with neutral oligosaccharides. Represents the desired product's primary structure [3]. |
| Basic Variants | Species with a higher isoelectric point (pI) than the main species [2]. | C-terminal unprocessed lysine, proline amidation, N-terminal pyroglutamate formation, and signal peptide residues [2] [4]. |
The biological impact of a charge variant depends on the type and location of the modification. Modifications in the Complementary Determining Region (CDR) can reduce antigen binding affinity and potency, making them product-related impurities [3]. Conversely, variants with modifications in non-critical regions (e.g., C-terminal lysine variants) that do not impact safety or efficacy are considered product-related substances [3]. A comprehensive characterization is therefore required to identify which charge variants are Critical Quality Attributes (CQAs) that must be controlled within strict limits [2] [4].
Analytical Techniques for Charge Variant Analysis
A combination of orthogonal analytical techniques is employed to separate, quantify, and characterize charge variants. The choice of method depends on the need for high-resolution separation, direct quantification, or hyphenation with mass spectrometry for identification.
Table 2: Key Analytical Techniques for Charge Variant Analysis
| Technique | Principle of Separation | Key Applications & Advantages |
|---|---|---|
| Cation Exchange Chromatography (CEX-HPLC) | Separates variants based on differences in net surface charge using a charged stationary phase and a salt gradient [3] [2]. | Industry standard for monitoring and fraction collection; provides robust quantification and preparative-scale isolation for further characterization [3]. |
| Capillary Zone Electrophoresis (CZE) | Separates variants based on their charge-to-size ratio in a capillary under an electric field [5] [6]. | High separation efficiency; emerging MS-compatible methods allow direct identification of variants [5] [6]. |
| imaged Capillary Isoelectric Focusing (icIEF) | Separates variants based on their intrinsic isoelectric point (pI) in a pH gradient [2]. | High-resolution separation and direct quantification of charge heterogeneity; primarily used for analytical testing [2]. |
A significant advancement in the field is the development of MS-compatible CZE methods. Traditional high-resolution CZE methods relied on non-volatile background electrolytes that were incompatible with mass spectrometry. Recent research has successfully implemented volatile electrolytes and specialized capillary coatings, enabling the direct coupling of high-efficiency separation with mass spectrometry (CZE-UV/MS) [5] [6]. This allows for the direct identification and quantitation of basic, acidic, and glycoforms of intact mAbs [6].
Detailed Experimental Protocols
Protocol for Ion-Exchange Chromatography (IEC) for Charge Variant Separation
This protocol describes the separation of charge variants using a strong cation-exchange chromatography column for analytical quantification [2].
I. Materials and Reagents
- Mobile Phase A: 25 mM N-(2-Acetamido)-2-aminoethanesulfonic acid (ACES) buffer, pH 7.0.
- Mobile Phase B: 150 mM Sodium Chloride in Mobile Phase A.
- Sample Buffer: Identical to Mobile Phase A.
- Column: Strong cation-exchange chromatography column (e.g., YMC-BioPro SP-F, 4.6 × 100 mm).
- HPLC System: Agilent 1200 HPLC system or equivalent.
II. Procedure
- Sample Preparation: Dilute the mAb sample to a concentration of 10 mg/mL using Mobile Phase A [2].
- System Setup: Equilibrate the column with 92% Mobile Phase A and 7% Mobile Phase B at a flow rate of 0.6 mL/minute. Maintain the column temperature at 36°C [2].
- Sample Injection: Inject 5 µL of the prepared sample (50 µg total) onto the column [2].
- Chromatographic Separation:
- Initiate the run with an isocratic hold at 7% Mobile Phase B for 4 minutes.
- Following the hold, apply a linear gradient to increase the concentration of Mobile Phase B from 7% to 15% over 10 minutes to elute the charge variants [2].
- Detection and Analysis: Monitor the eluent using UV detection (e.g., 280 nm). Identify the main peak, acidic variants (typically earlier eluting), and basic variants (typically later eluting).
Protocol for Capillary Zone Electrophoresis-Mass Spectrometry (CZE-UV/MS)
This protocol outlines a generic CZE-MS method for high-resolution charge variant analysis using a cationic capillary coating and an acidic, volatile background electrolyte [5].
I. Materials and Reagents
- Capillary Coating: Successive multiple ionic-polymer layer (SMIL) coating based on diethylaminoethyl–dextran (DEAED) and poly(sodium styrene sulfonate) (PSS) [5].
- Background Electrolyte (BGE): 50 mM acetic acid, adjusted to pH 5.0 with ammonium hydroxide [6].
- Capillary: Fused silica capillary with SMIL coating.
- Antibody Solution: 1 mg/mL mAb solution prepared in the BGE [5].
II. Procedure
- Capillary Preparation: If using a SMIL coating, flush a new or used capillary with 1 M NaOH for 10 min, followed by ultrapure water for 5 min and a 20 mM HEPES solution (pH 7.4) for 10 min. Build the SMIL coating by alternately flushing with polycation and polyanion solutions until five layers are reached [5].
- Equilibration: Before each analysis, flush the capillary with BGE for 10 minutes [5].
- Sample Injection: Hydrodynamically inject the mAb sample using 40 mbar for 5 seconds [5].
- Separation: Apply a separation voltage of -10 kV (for SMIL coating). The reversed electroosmotic flow enables the separation of variants with slightly different mobilities [5].
- MS Coupling and Detection: Couple the CZE system to the mass spectrometer using a low-flow sheath liquid interface (e.g., nanoCEasy). The volatile BGE at pH 5.0 allows for efficient electrospray ionization and mass spectrometric detection of the separated intact mAb charge variants [5] [6].
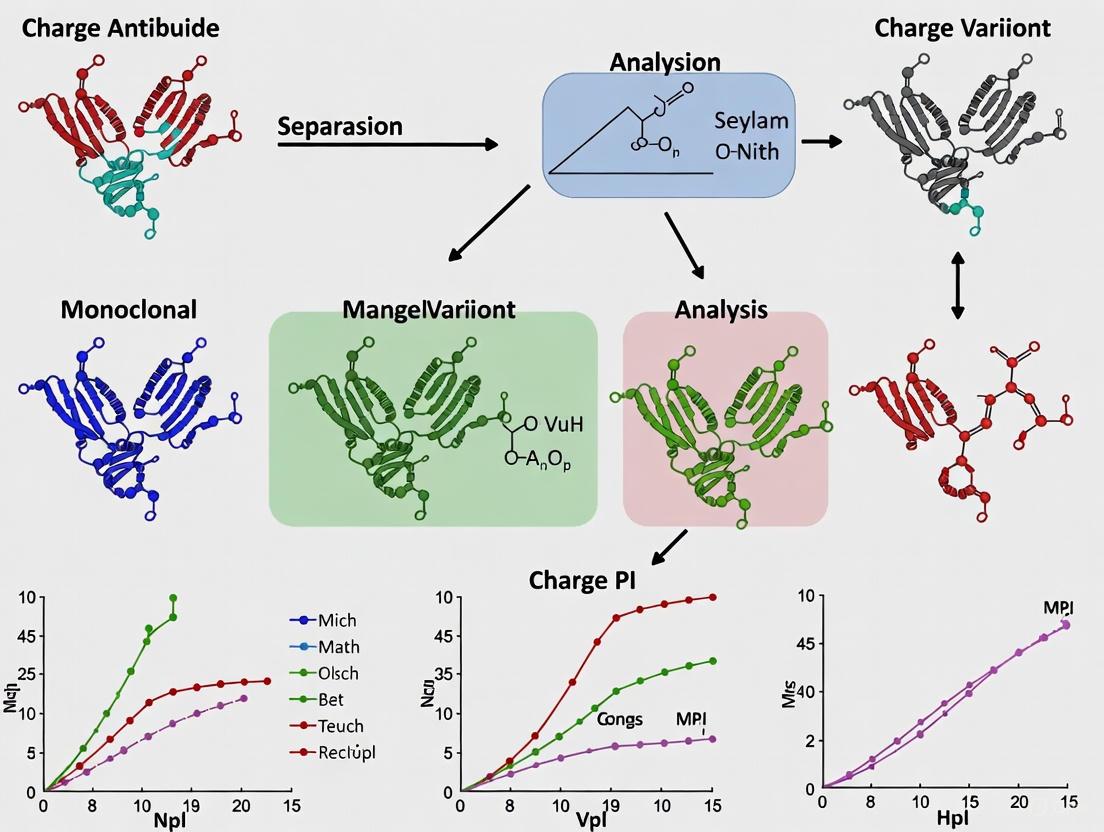
Workflow Visualization
The following diagram illustrates the logical workflow for the characterization of charge variants in therapeutic monoclonal antibodies, from initial analysis to final classification.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful charge variant analysis relies on a suite of specialized reagents and materials. The following table details key items essential for experiments in this field.
Table 3: Essential Research Reagents and Materials for Charge Variant Analysis
| Item | Function / Application |
|---|---|
| Strong Cation-Exchange Column (e.g., YMC-BioPro SP-F) | Stationary phase for separating charge variants based on surface charge differences using HPLC [2]. |
| ACES Buffer (25 mM, pH 7.0) | Volatile buffer system used as mobile phase in IEC to maintain a stable pH during separation without interfering with detection [2]. |
| SMIL Capillary Coating (DEAED-PSS) | A cationic capillary coating that generates a controlled electroosmotic flow (EOF) for high-resolution CZE separations under MS-compatible conditions [5]. |
| Volatile BGE (50 mM Ammonium Acetate, pH 5.0) | An acidic, volatile background electrolyte for CZE that enables direct hyphenation with ESI-MS by not leaving residues that disrupt ionization [6]. |
| Carboxypeptidase B (CPB) | Enzyme used under native conditions to enzymatically remove C-terminal lysine residues from mAbs, helping to characterize their contribution to basic species [3]. |
| Sialidase | Enzyme used under native conditions to remove sialic acid residues from glycans, helping to characterize their contribution to acidic species [3]. |
Key Post-Translational Modifications (PTMs) Driving Charge Variation
In the development of monoclonal antibody (mAb) therapeutics and related modalities, charge variant analysis is a critical component for ensuring product quality, consistency, and efficacy. Therapeutic proteins are inherently heterogeneous due to chemical and enzymatic post-translational modifications (PTMs) that occur during manufacturing, purification, and storage [2]. These modifications alter the protein's overall surface charge distribution, creating charge variants—species with isoelectric points (pI) lower than the main product (acidic variants) or higher (basic variants) [2]. As many of these PTMs can impact therapeutic potency, stability, and immunogenicity, they are monitored as Critical Quality Attributes (CQAs) requiring thorough characterization throughout the drug development lifecycle [7] [8].
Key PTMs Driving Charge Heterogeneity
The following table summarizes the primary PTMs responsible for charge variation, their molecular consequences, and their typical impact on chromatographic elution.
Table 1: Key Post-Translational Modifications Driving Charge Variation in Monoclonal Antibodies
| Modification Type | Specific PTM | Molecular Effect | Impact on Net Charge | Chromatographic Region |
|---|---|---|---|---|
| Deamidation | Asparagine (Asn) to Aspartate/Isoaspartate | Addition of carboxylic acid group; potential succinimide intermediate [7] | Increase in negative charge | Acidic [2] |
| C-terminal Lysine | Lysine clipping (removal) | Removal of a primary amine group [9] | Reduction in positive charge (more negative) | Acidic [2] |
| Glycation | Lysine side chain modification by glucose | Schiff base formation; neutral mass addition can mask positive charge [2] | Reduction in positive charge | Acidic [2] |
| Oxidation | Methionine to Methionine Sulfoxide | Addition of oxygen; can alter local structure [7] | Can elute in basic region [2] | Basic/Acidic |
| N-linked Glycosylation | Sialylation (addition of sialic acid) | Addition of one or more negatively charged sugar residues [2] | Increase in negative charge | Acidic [2] |
| Proline Amidation | C-terminal proline amidation | Neutral modification [2] | Can contribute to basic character | Basic [2] |
| Succinimide Formation | Cyclization of Asparagine or Aspartate | Neutral intermediate prone to hydrolysis [7] | Can lead to acidic variants upon decomposition | Acidic [7] |
| Isomerization | Aspartate to Isoaspartate | Structural rearrangement of aspartic acid [7] | Can alter local charge environment | Acidic [7] |
Characterizing these variants presents a significant analytical challenge. A comprehensive study of a therapeutic mAb1 revealed that while basic variants (e.g., unprocessed lysine, proline amidation) could be fully accounted for, nearly one-third of the acidic variants remained unidentified in initial characterization studies, highlighting the complexity of fully mapping the charge variant landscape [2].
Analytical Techniques for Charge Variant Characterization
A multi-technique approach is essential for separating, isolating, and identifying charge variants. The following workflow diagram illustrates the integrated strategy employed for comprehensive characterization.
Diagram 1: Charge Variant Characterization Workflow
Separation and Quantification Techniques
- Ion-Exchange Chromatography (IEC): The most widely used method, particularly cation-exchange chromatography (CEX), separates variants based on their differential interaction with a charged stationary phase under controlled pH and ionic strength gradients [2] [8]. Modern methods employ volatile buffers (e.g., ammonium acetate) to enable direct coupling with mass spectrometry [7] [10].
- Capillary Electrophoresis Techniques: Capillary Zone Electrophoresis (CZE) and capillary isoelectric focusing (cIEF) offer high-resolution separation based on electrophoretic mobility or pI, respectively [11] [12]. CZE-UV has proven effective for rapid biosimilarity assessments by comparing charge variant profiles of innovator and biosimilar products like infliximab [12].
Identification and Characterization Techniques
- Native Mass Spectrometry (Native MS): The coupling of CEX using volatile buffers with native MS allows for the direct determination of molecular weights associated with different charge variants, enabling rapid identification of modifications like deamidation, oxidation, and glycosylation without extensive sample preparation [7] [10].
- Peptide Mapping: This bottom-up approach involves digesting the protein (typically with trypsin) and analyzing the peptides by LC-MS/MS. It is the gold standard for pinpointing the exact location and quantity of PTMs at the amino acid residue level, confirming modifications suggested by intact analyses [7] [13].
- Limited Digestion: For complex molecules like bispecific antibodies, limited digestion with enzymes such as Lys-C can cleave the molecule into defined large fragments (e.g., individual domains), allowing for chromatographic resolution and MS analysis to determine PTMs associated with specific functional regions [7].
Detailed Experimental Protocol: CEX-MS for Intact Charge Variant Analysis
This protocol describes the use of pH-gradient cation-exchange chromatography coupled online with native mass spectrometry for the intact analysis of charge variants in a bispecific antigen-binding protein (BsABP), as detailed by [7] [10].
Materials and Reagents
Table 2: Research Reagent Solutions for CEX-MS Analysis
| Item | Specification | Function / Purpose |
|---|---|---|
| Cation-Exchange Column | YMC-BioPro SP-F (100 mm × 4.6 mm, 5 μm) or equivalent [7] | Separation of charge variants based on surface charge differences. |
| Ammonium Acetate | MS-grade, for volatile buffer preparation [7] | Provides buffering capacity and ionic strength for separation while being compatible with MS detection. |
| Ammonium Hydroxide | For pH adjustment [7] | Used to prepare basic mobile phase (Solvent B). |
| Formic Acid | For pH adjustment [7] | Used to prepare acidic mobile phase (Solvent A). |
| Therapeutic Protein | mAb or BsABP, formulated at low concentration (e.g., ~2 mg/mL) [7] | The analyte of interest for charge variant characterization. |
Instrumentation and Method Conditions
- LC System: Waters UPLC or Alliance iS Bio HPLC System equipped with a binary pump and column heater. For high-pH work, a system with a corrosion-resistant high-pH kit is recommended [14].
- Mass Spectrometer: High-resolution mass spectrometer equipped with a native ESI source (e.g., Biopharma Orbitrap Q-Exactive Plus) [7].
- Mobile Phase Preparation:
- Chromatographic Conditions:
- Mass Spectrometry Conditions:
Procedure
- System Equilibration: Equilibrate the CEX column with Solvent A (100%) for at least 15 minutes or until a stable baseline is achieved at the specified flow rate.
- Sample Preparation: If necessary, dilute the protein sample (e.g., BsABP drug substance) into Solvent A to the desired concentration. Centrifuge to remove particulates.
- Sample Injection: Inject the prepared sample (30 μg) onto the column.
- Chromatographic Separation: Initiate the pH gradient method as described. Monitor the elution of charge variants by UV at 280 nm.
- Online MS Detection: As variants elute from the column, they are directly introduced into the mass spectrometer. The volatile ammonium acetate buffers allow for efficient desolvation and ionization under native conditions, producing ions with lower charge states that preserve non-covalent interactions.
- Data Analysis:
- Chromatographic Data: Integrate the UV chromatogram to quantify the relative proportions of acidic, main, and basic variant peaks.
- Mass Spectrometric Data: Deconvolute the native mass spectra for each eluting peak to determine the molecular weights of the proteoforms. Identify modifications by comparing the observed mass shifts with theoretical values for common PTMs (e.g., +1 Da for deamidation, +16 Da for oxidation).
The comprehensive characterization of charge variants driven by PTMs is a non-negotiable requirement in the development of biotherapeutics. The integration of advanced separation techniques like CEX and CZE with powerful detection methods such as native MS and peptide mapping provides a robust framework for identifying and quantifying these critical quality attributes. The experimental workflow and detailed protocol outlined herein enable researchers to elucidate the complex charge heterogeneity of monoclonal antibodies and newer modalities, ensuring the delivery of safe, effective, and high-quality biologic drugs to patients.
Deamidation, Isomerization, and Sialylation as Major Contributors to Acidic Variants
Charge heterogeneity in monoclonal antibodies (mAbs) represents a significant challenge in biopharmaceutical development, with acidic variants being a primary focus due to their potential impact on drug stability, efficacy, and safety. This application note delineates the principal molecular drivers of acidic variant formation—deamidation, isomerization, and sialylation—within the broader context of charge variant analysis. We provide detailed experimental protocols for the characterization and quantification of these critical quality attributes (CQAs), supported by structured data presentation and workflow visualizations. The methodologies outlined herein, including imaged capillary isoelectric focusing (icIEF) and cation exchange chromatography (CEX), offer robust analytical frameworks for researchers and drug development professionals to monitor and control charge heterogeneity throughout the product lifecycle, ensuring consistent biotherapeutic quality.
Monoclonal antibodies constitute a dominant class of biotherapeutics for treating oncological, inflammatory, and autoimmune diseases [15]. Their structural complexity, however, renders them susceptible to various post-translational modifications (PTMs) during production, purification, and storage, leading to significant product heterogeneity [15] [16]. Charge-based heterogeneity is a key CQA scrutinized by regulatory authorities, as it can influence biological activity, pharmacokinetics, and storage stability [15] [16] [17].
Acidic variants, which exhibit a more negative charge and lower isoelectric point (pI) than the main species, are of particular concern [18] [19]. The formation of these variants is predominantly driven by specific chemical modifications: deamidation of asparagine residues, isomerization of aspartic acid residues, and sialylation of N-linked glycans on the Fc region [18] [19] [3]. Thorough characterization of these variants is therefore essential for demonstrating batch-to-batch consistency, supporting process validation, and ensuring final product quality [3] [20]. This document provides detailed protocols and analytical strategies for elucidating the role and impact of these major contributors to acidic charge variants.
Mechanisms and Impact of Major Acidic Variants
The following table summarizes the key attributes of the three major contributors to acidic variants in mAbs.
Table 1: Key Characteristics of Major Acidic Variant Contributors
| Modification | Chemical Change | Net Charge Effect | Primary Location | Potential Impact on mAb |
|---|---|---|---|---|
| Deamidation [16] [17] | Conversion of Asn to Asp/isoAsp | Gain of negative charge | Complementarity-determining regions (CDRs), Fc | Reduced binding affinity, altered potency, increased aggregation propensity |
| Isomerization [17] [21] | Conversion of Asp to isoAsp | Altered local charge distribution | CDRs | Reduced binding affinity and biological activity |
| Sialylation [22] [19] | Addition of sialic acid to glycan chains | Gain of negative charge | Fc glycans | Potential modulation of immune effector functions (e.g., ADCC) |
Deamidation
Deamidation is a common chemical degradation pathway where the neutral side chain of asparagine (Asn) is converted to a negatively charged aspartic acid (Asp) or isoaspartic acid (isoAsp) residue [16] [17]. This process is highly dependent on pH, temperature, and the protein's primary structure, particularly the sequence following the Asn residue [17]. When deamidation occurs in the CDRs, it can directly interfere with antigen binding, reducing the potency and efficacy of the therapeutic mAb [16] [3]. Furthermore, deamidation has been linked to reduced colloidal stability and can enhance aggregation propensity, especially under acidic conditions [17].
Isomerization
Isomerization involves the conversion of aspartic acid (Asp) to its structural isomer, isoaspartic acid (isoAsp), via a succinimide intermediate [17] [21]. This reaction can occur spontaneously under acidic conditions. While the net charge change may be subtle, isomerization can induce significant conformational shifts in the mAb structure [21]. These small conformational changes can alter the local surface charge distribution, which is detectable by chromatographic methods like CEX, and can critically impair antigen-binding affinity when located in the CDRs [17] [21].
Sialylation
Sialylation is an enzymatic PTM wherein sialic acid residues are added to the terminal end of N-linked glycans on the mAb's Fc region [22] [19]. Each sialic acid introduces a negative charge. For mAbs produced in Chinese Hamster Ovary (CHO) cells, sialic acids are exclusively linked in an α2,3 orientation [22]. The impact of Fc sialylation on antibody-dependent cellular cytotoxicity (ADCC) has been debated, with some studies showing little to no effect [22]. Nonetheless, because it significantly alters the molecule's pI, it is a major contributor to the acidic variant profile monitored during quality control.
Analytical Methodologies and Experimental Protocols
Research Reagent Solutions
The following table lists essential reagents and materials required for the experimental workflows described in this note.
Table 2: Key Research Reagent Solutions for Charge Variant Analysis
| Reagent/Material | Function/Application | Example Usage |
|---|---|---|
| icIEF Instrumentation | High-resolution separation and quantification of charge variants based on pI. | Platform charge heterogeneity profiling for QC [15]. |
| Cation Exchange (CEX) Column | Chromatographic separation of charge variants for purity assessment and fraction collection. | Isolation of acidic, main, and basic peaks for further characterization [3]. |
| Sialidase (Neuraminidase) | Enzymatic removal of terminal sialic acid residues from glycan chains. | Investigating the contribution of sialylation to acidic peaks [22] [21]. |
| Carboxypeptidase B (CPB) | Enzymatic cleavage of C-terminal lysine residues. | Differentiating C-terminal lysine variants from other basic species [3]. |
| LC-MS/MS System | High-sensitivity identification and precise localization of PTMs (e.g., deamidation, isomerization). | Peptide mapping for definitive variant characterization [3] [20]. |
| Carrier Ampholytes (CAs) | Create a stable pH gradient within the capillary for icIEF separation. | Essential additive for icIEF method development [15]. |
Protocol 1: Charge Variant Profiling via Imaged Capillary Isoelectric Focusing (icIEF)
Principle: icIEF separates protein charge variants based on their isoelectric point (pI) within a coated capillary. Under an electric field, variants migrate until they reach a pH zone where their net charge is zero (pI), forming focused bands that are detected by a CCD camera [15].
Procedure:
- Sample Preparation: Dilute the mAb drug substance to a concentration of 0.5-1 mg/mL in a solution containing 0.35% methylcellulose, 2-4% carrier ampholytes (pH 3-10 gradient), and 0.5% pI markers (e.g., pI 7.6 and 9.5) [15]. Additives such as 1-2 M urea can be included to improve solubility and resolution.
- Focusing: Introduce the sample mixture into the icIEF capillary. Apply a voltage of 3.0-5.0 kV for 5-10 minutes at a defined temperature (e.g., 20°C) until the current stabilizes at a minimum, indicating successful focusing [15].
- Detection & Analysis: Image the entire capillary length using a UV detector (e.g., 280 nm). The resulting electropherogram will display peaks corresponding to acidic variants, the main species, and basic variants. Integrate peak areas to calculate the percentage distribution of each group [15].
Protocol 2: Isolation and Characterization of Acidic Variants
Principle: Cation exchange chromatography (CEX) is used to separate and collect acidic variant fractions at a semi-preparative scale. The isolated fractions are then characterized using orthogonal techniques to identify specific modifications [3].
Procedure:
- Preparative CEX Separation:
- Transfer an analytical CEX-HPLC method to a semi-preparative scale, optimizing the salt gradient for optimal resolution of acidic peaks [3].
- Perform multiple injections of the mAb sample (e.g., 10-50 mg total) and collect the early-eluting peaks corresponding to the acidic variants. Pool the fractions from multiple runs.
- As a control, also collect the main peak fraction.
- Purity Assessment: Analyze the collected fractions using the original analytical CEX-HPLC method. Overlay the chromatograms with the unfractionated sample to confirm enrichment and purity (>80% is desirable) [3].
- Enzymatic Treatment for Characterization:
- To Probe Sialylation: Incubate an aliquot of the isolated acidic fraction and the main fraction with sialidase (e.g., 5 U/mL) at 37°C for 1 hour [22] [21]. Re-analyze the treated samples by CEX or icIEF. A reduction in the acidic peak area suggests that sialylation contributes to that variant [21].
- For Comprehensive PTM Identification: Subject the isolated fractions to reduction and enzymatic digestion (e.g., with trypsin). Analyze the resulting peptides using LC-MS/MS with peptide mapping. This allows for the precise identification of deamidated (mass shift +0.984 Da) or isomerized peptides, localizing the modifications to specific residues in the sequence [3] [20].
Diagram 1: Acidic Variant Characterization Workflow. This diagram outlines the key steps for isolating and characterizing acidic variants, from initial fractionation to final data analysis.
Data Presentation and Interpretation
Quantitative Analysis of Charge Variants
The following table provides an example dataset illustrating the typical distribution of charge variants and the impact of enzymatic treatments on a hypothetical IgG1 mAb.
Table 3: Example Charge Variant Distribution of an IgG1 mAb Before and After Enzymatic Treatment
| Sample Condition | Acidic Variants (%) | Main Peak (%) | Basic Variants (%) | Notes |
|---|---|---|---|---|
| Starting Material | 24.5 | 68.2 | 7.3 | Baseline profile [19] |
| After Sialidase Treatment | 18.1 | 74.6 | 7.3 | Suggests ~6.4% of acidic variants are sialylated |
| After CPB Treatment | 24.5 | 74.9 | 0.6 | Confirms basic variants are primarily C-terminal Lysine |
Case Study: Investigating Conformational Variants
Recent studies indicate that not all acidic variants can be explained by simple chemical modifications. Some arise from small conformational changes that alter the surface charge distribution without modifying the primary sequence [21]. This can be investigated by subjecting the main peak fraction to a controlled refolding process. The generation of new acidic peaks upon refolding, as detected by CEX, provides evidence for the presence of conformational variants, which may have been misfolded during production or storage [21].
Diagram 2: Conformational Variant Analysis. A refolding experiment can generate acidic variants, revealing conformational differences that contribute to charge heterogeneity.
Deamidation, isomerization, and sialylation are established as major biochemical drivers of acidic variant formation in therapeutic mAbs. Effectively monitoring and controlling these CQAs is a regulatory expectation and a crucial aspect of ensuring product quality. The integrated analytical strategies presented here—combining high-resolution separation techniques like icIEF and CEX with orthogonal characterization methods such as enzymatic digestion and LC-MS/MS—provide a comprehensive toolkit for researchers. By implementing these detailed protocols, scientists can gain deep insights into the root causes of charge heterogeneity, enabling robust process development, rigorous quality control, and the delivery of safe and efficacious biotherapeutic products.
In the development and manufacturing of therapeutic monoclonal antibodies (mAbs), charge heterogeneity is a critical quality attribute that must be thoroughly characterized to ensure product consistency, efficacy, and safety [19] [3]. Charge variants are typically categorized into acidic, main, and basic species based on their elution profiles in ion-exchange chromatography [23]. Basic variants, which elute later than the main peak in cation-exchange chromatography (CEX), are of particular interest due to their potential impacts on biological activity and pharmacokinetics [19] [24]. Among the various post-translational modifications (PTMs) that contribute to basic variant formation, C-terminal lysine, succinimide formation, and oxidation represent three major sources with distinct chemical origins and functional consequences [19] [25] [23]. Understanding these modifications is essential for establishing meaningful control strategies throughout the biopharmaceutical development lifecycle.
Structural Basis and Functional Impact of Major Basic Variants
C-terminal Lysine
The presence of C-terminal lysine residues on the heavy chains of monoclonal antibodies constitutes a common source of basic charge variants [25] [3]. During antibody production in mammalian expression systems such as Chinese hamster ovary (CHO) cells, carboxypeptidases in the culture medium may incompletely cleave the C-terminal lysine residues, resulting in antibody populations with 0, 1, or 2 lysine residues [25]. Each retained lysine contributes an additional positive charge at neutral pH, increasing the antibody's isoelectric point (pI) and causing later elution in CEX chromatography [19] [23]. Although C-terminal lysine variants are generally not expected to affect safety or efficacy since these regions are highly exposed and not part of ligand binding sites [3], they can complicate charge variant profiles and must be monitored for batch-to-batch consistency [19].
Succinimide Formation
Succinimide intermediate formation represents another important mechanism for generating basic variants through the cyclization of aspartic acid or asparagine residues [23]. This process typically occurs via deamidation of asparagine or isomerization of aspartic acid, resulting in the formation of a cyclic imide intermediate that lacks the negative charge of the parent residue [19] [23]. The loss of this negative charge effectively increases the pI of the antibody molecule, leading to its classification as a basic variant [19]. Succinimide intermediates are particularly noteworthy for their instability under typical denaturation, reduction, alkylation, and enzymatic digestion conditions used for LC-MS analysis, making their characterization challenging [3]. These intermediates can subsequently hydrolyze to either aspartic acid or isoaspartic acid, the latter of which has been associated with potential alterations in biological activity, especially when occurring in complementarity-determining regions (CDRs) [3].
Oxidation
Amino acid oxidation, particularly of methionine, tryptophan, and cysteine residues, constitutes a third major pathway for basic variant formation [19] [23]. Oxidation introduces more hydrophilic groups and can cause conformational changes that alter surface charge distribution, potentially increasing pI and contributing to the basic variant profile [19] [23]. The impact of oxidation on biological function depends largely on the location of the modified residue. Oxidation in the Fc region, particularly of methionine residues, has been shown to affect neonatal Fc receptor (FcRn) binding, which may influence the antibody's serum half-life [3]. When oxidation occurs in the Fab region, especially within CDRs, it can directly impact antigen binding affinity and reduce potency [3].
Table 1: Characteristics of Major Basic Variant Modifications in Monoclonal Antibodies
| Modification | Chemical Basis | Charge Effect | Potential Functional Impact | Analytical Detection Methods |
|---|---|---|---|---|
| C-terminal Lysine | Incomplete cleavage by carboxypeptidases | Increases positive charge (+1 per Lys) | Generally minimal impact on safety/efficacy [3] | CEX, icIEF, peptide mapping, intact mass analysis [25] [3] |
| Succinimide Formation | Cyclization of Asp or Asn residues | Loss of negative charge | Potential alteration in activity if in CDRs [3] | Peptide mapping (with special handling) [3] |
| Oxidation | Addition of oxygen to Met, Trp, Cys | Conformational change affecting surface charge | Altered FcRn binding (half-life) or antigen binding (potency) [3] | Peptide mapping, intact mass analysis, CEX [3] |
Quantitative Assessment of Basic Variants
Comprehensive characterization of therapeutic mAbs requires quantitative assessment of the various basic variant species present in drug substances and products. Research on multiple therapeutic antibodies has revealed substantial variation in the relative abundance of different basic species, with some modifications demonstrating particularly high prevalence in certain products.
Table 2: Relative Abundances of Basic Variants in Characterized Monoclonal Antibodies
| mAb Identifier | C-terminal Lysine | C-terminal Amidation | Succinimide | Oxidation | Other Basic Variants | Citation |
|---|---|---|---|---|---|---|
| ch14.18 | Present (specific % not reported) | Not reported | Identified as source | Not quantified | N-terminal pyroglutamate from Glu, different glycoforms | [24] |
| mAb1 (Genentech) | Not predominant | Not reported | Not predominant | Detected in basic region | Proline amidation, signal peptides (account for ~93% of basics) | [26] |
| General IgG1 | ~12% of total charge variants (average) | Detected in 8 of 12 mAbs studied | Identified as source | Listed as source of basic variants | N-terminal pyroglutamate from Glu, disulfide-mediated modifications | [19] [25] |
Experimental Protocols for Basic Variant Characterization
Isolation of Basic Charge Variants
Principle: Basic charge variants are separated from acidic and main species using scalable cation-exchange chromatography (CEX) under conditions that resolve variants based on their surface charge differences [19] [24].
Procedure:
- Equilibrate a strong cation-exchange column (e.g., YMC-BioPro SP-F, ProPac WCX-10, or MAbPac SCX-10) with mobile phase A (25 mM ACES buffer, pH 7.0, or 20-25 mM sodium phosphate buffer) [26] [27].
- Dilute the mAb sample to 10-30 mg/mL with mobile phase A [19] [26].
- Inject the sample and elute using either:
- Monitor elution at 280 nm and collect the late-eluting peaks corresponding to basic variants [19] [3].
- Concentrate and buffer-exchange the collected fractions using centrifugal concentrators [3].
- Verify the purity of isolated fractions by re-analyzing an aliquot with analytical CEX [3].
Critical Considerations:
- For mAbs with C-terminal lysine variants, treatment with carboxypeptidase B (CPB) before fraction collection can help enrich other basic variants by removing the C-terminal lysine contribution [3].
- Maintain consistent pH and temperature throughout the process to prevent artificial variant generation [3].
- Use volatile salts like ammonium acetate when the collected fractions are intended for mass spectrometric analysis [28].
Enzymatic Treatment for C-terminal Lysine Detection
Principle: Carboxypeptidase B (CPB) specifically cleaves C-terminal lysine and arginine residues, allowing confirmation of C-terminal lysine contribution to basic variants [29] [3].
Procedure:
- Prepare the mAb sample at 1-2 mg/mL in appropriate buffer (e.g., 20 mM Tris-HCl, pH 7.5) [3].
- Add CPB at an enzyme-to-substrate ratio of 1:50 to 1:100 (w/w) [3].
- Incubate at 37°C for 30-60 minutes [3].
- Stop the reaction by acidification or immediate analysis.
- Analyze the digested sample alongside untreated control using CEX or icIEF.
- The reduction in basic peak area indicates the proportion attributable to C-terminal lysine [3].
Peptide Mapping for Succinimide and Oxidation Detection
Principle: Tryptic peptide mapping with LC-MS/MS identifies specific modification sites and differentiates between succinimide intermediates and their hydrolysis products [3].
Procedure:
- Denature the isolated basic variant fraction in guanidine hydrochloride or SDS [3].
- Reduce disulfide bonds with dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) [23] [3].
- Alkylate cysteine residues with iodoacetamide [3].
- Digest with trypsin (1:20-1:50 enzyme-to-substrate ratio) at 37°C for 4-16 hours [25] [3].
- Analyze the resulting peptides by reversed-phase LC-MS/MS.
- Identify succinimide-containing peptides by:
- Mass shift of -17 Da (for asparagine) or -18 Da (for aspartic acid) [3]
- Characteristic fragmentation patterns in MS/MS spectra
- Identify oxidized peptides by:
- Mass shifts of +16 Da (single oxidation) or +32 Da (double oxidation)
- Specific residues (methionine, tryptophan, cysteine) [3]
Critical Considerations:
- Succinimide intermediates are unstable under standard digestion conditions and may require special handling (e.g., acidic pH or lower temperature) to preserve the modification [3].
- Use parallel digestion with Asp-N protease when trypsin cleavage is compromised by modifications [25].
- Include proper controls to distinguish between process-related and artificially induced modifications [3].
Analytical Workflow for Basic Variant Characterization
The following diagram illustrates the comprehensive workflow for characterizing basic variants in therapeutic monoclonal antibodies:
Research Reagent Solutions
Successful characterization of basic variants requires carefully selected reagents and methodologies. The following table outlines essential solutions for comprehensive analysis:
Table 3: Essential Research Reagents for Basic Variant Characterization
| Reagent/Instrument | Function | Application Notes |
|---|---|---|
| Strong Cation-Exchange Columns (ProPac WCX-10, MAbPac SCX-10, YMC-BioPro SP-F) | Separation of charge variants based on surface charge differences | Use pH gradients with volatile salts (ammonium acetate) for MS compatibility [28] [27] |
| Carboxypeptidase B (CPB) | Selective removal of C-terminal lysine residues | Confirm C-terminal lysine contribution to basic variants; use native conditions to preserve structure [29] [3] |
| Trypsin/Asp-N Protease | Proteolytic digestion for peptide mapping | Identify modification sites; Asp-N useful when trypsin cleavage compromised [25] [3] |
| UHPLC Systems (Vanquish Flex UHPLC) | High-resolution separation with minimal extracolumn volume | Essential for reproducible charge variant profiling; biocompatible systems prevent surface adsorption [27] |
| MS-Compatible Ampholytes | cIEF focusing without MS interference | Enable direct coupling of cIEF to mass spectrometry for variant identification [28] |
| Surface Plasmon Resonance (Biacore systems) | Binding affinity measurements for FcRn and antigens | Assess functional impact of basic variants on target engagement and FcRn binding [24] |
The comprehensive characterization of C-terminal lysine, succinimide intermediates, and oxidation products as sources of basic variants is essential for ensuring the quality, efficacy, and safety of therapeutic monoclonal antibodies. Through the implementation of robust analytical workflows incorporating orthogonal methodologies, researchers can accurately identify and quantify these modifications, understand their functional consequences, and establish appropriate control strategies during biopharmaceutical development. The continued refinement of these characterization approaches will further enhance our ability to manage charge heterogeneity in therapeutic antibodies, ultimately leading to improved product quality and consistency.
The Critical Link Between Charge Variants and Drug Safety, Efficacy, and Stability
Charge heterogeneity in monoclonal antibodies (mAbs) refers to the presence of molecular variants that possess distinct net surface charges, typically categorized into acidic species, main species, and basic species. [18] This heterogeneity represents a substantial challenge in ensuring consistent quality of biopharmaceuticals because charge variants can significantly impact therapeutic efficacy, product stability, and patient safety. [18] [30] Regulatory agencies including the FDA and EMA have established strict guidelines requiring manufacturers to closely monitor and control charge variation during process development and optimization. [18]
The presence of charge variants stems primarily from post-translational modifications (PTMs) that occur during biomanufacturing, storage, or even in vivo. [18] [12] These modifications include deamidation, oxidation, glycosylation variations, C-terminal lysine processing, and many other chemical alterations that change the molecule's isoelectric point (pI). [30] The distribution of these charge variants is highly sensitive to process conditions, making charge variant profiling a critical indicator of process robustness and product consistency. [30] Failure to maintain consistent charge profiles can substantially affect clinical development timelines and jeopardize batch disposition decisions. [30]
Root Causes and Molecular Mechanisms of Charge Heterogeneity
Primary Modifications Leading to Acidic Variants
Acidic variants, which possess a more negative charge than the main species, predominantly arise from specific chemical modifications that either add negative charges or mask positive ones. [18] The most prevalent modifications contributing to acidic species include:
- Deamidation of asparagine residues converts asparagine to aspartic acid or isoaspartic acid, adding negative charges to the molecule. This process is accelerated under elevated pH and temperature conditions and is considered a primary driver of acidic variant formation. [18] [30]
- Sialylation of N-glycans involves the enzymatic addition of sialic acids to carbohydrate structures, increasing negative charge. [18]
- Glycation of lysine residues occurs through non-enzymatic reactions with reducing sugars, which masks the positive charges on lysine side chains. [18] [30]
- Oxidation of methionine and tryptophan residues can lead to conformational changes that affect chromatographic behavior, even when they don't directly modify the overall charge. [18]
Primary Modifications Leading to Basic Variants
Basic variants, characterized by a higher positive charge than the main species, typically result from different sets of modifications:
- Incomplete removal of C-terminal lysine residues represents a major source of basic variants, as each retained lysine adds a positive charge. [18] [30] [31]
- Incomplete formation of N-terminal pyroglutamate from glutamine or glutamate leaves positive charges unneutralized. [18]
- Succinimide formation temporarily neutralizes negative charges, resulting in a more basic species. [18] [31]
- C-terminal amidation through enzymatic processing adds positive charges to the molecule. [18]
Table 1: Comprehensive Classification of Charge Variant Modifications
| Variant Type | Modification | Chemical Change | Effect on Charge | Prevalence |
|---|---|---|---|---|
| Acidic | Deamidation (Asn → Asp/isoAsp) | Conversion of neutral amide to acidic carboxyl group | Adds negative charge | Very Common [30] |
| Sialylation | Addition of sialic acids to glycans | Adds negative charge | Common [18] | |
| Glycation | Covalent modification of Lys by reducing sugars | Masks positive charge | Common [30] | |
| Oxidation (Met, Trp) | Addition of oxygen to sulfur/indole rings | Conformational changes affecting charge display | Common [18] | |
| Unformed disulfide bonds | Free thiol groups | Can affect charge distribution | Less Common [30] | |
| Basic | Incomplete C-terminal Lys removal | Retention of Lys residues | Adds positive charge | Very Common [18] [30] |
| Incomplete N-terminal pyroGlu formation | Retention of Glu/Gln at N-terminus | Retains positive charge | Common [18] | |
| Succinimide formation | Cyclization of Asp/Asn | Neutralizes negative charge temporarily | Common [18] [31] | |
| C-terminal amidation | Addition of amide group | Adds positive charge | Less Common [18] |
Impact of Charge Variants on Therapeutic Profile
Effects on Drug Safety and Immunogenicity
Charge variants can significantly influence the safety profile of therapeutic mAbs through multiple mechanisms. Heterogeneous charge profiles may increase the risk of immunogenic responses if structural modifications create novel epitopes that the immune system recognizes as foreign. [18] Although many charge variants are also found in endogenous human IgGs, which may alleviate some safety concerns, the unpredictable nature of immune responses necessitates careful monitoring. [30] Modifications that affect protein folding can lead to increased aggregation propensity, potentially enhancing immunogenicity and posing direct safety risks to patients. [18]
Effects on Biological Activity and Efficacy
The therapeutic efficacy of mAbs can be substantially compromised by specific charge-sensitive modifications, particularly when they occur in critical functional regions:
- Complementarity-determining regions (CDRs) containing deamidated asparagine residues have demonstrated decreased binding affinity to targets. For instance, a specific Asn deamidation was correlated with disruption of IgG1 binding to the Fc gamma receptor, directly leading to loss of antibody-dependent cell-mediated cytotoxicity (ADCC) activity. [12] [30]
- Succinimide intermediate formation and deamidation in CDRs have been successfully linked to decreased binding activity of mAbs to their targets. [12]
- While some modifications like C-terminal lysine variants typically show minimal impact on potency, others—especially those in critical functional domains—can significantly compromise therapeutic efficacy. [30] [31]
Effects on Stability and Pharmacokinetics
Charge heterogeneity directly influences product stability and pharmacokinetic properties:
- Acidic variants are often linked to degradation pathways (deamidation, oxidation) and may demonstrate increased aggregation propensity or fragmentation. [18]
- Substantial levels of methionine oxidation have been shown to cause shorter half-life in circulation, though this modification may not necessarily affect pI. [30]
- While some studies have shown minimal pharmacokinetic differences between isolated acidic and basic species for specific mAbs, substantial pI differences have been suspected to affect biodistribution. [30]
The following diagram illustrates the interconnected relationship between process parameters, charge variants, and critical quality attributes:
Analytical Methods for Charge Variant Characterization
Established Separation Techniques
Robust analytical methods are essential for comprehensive characterization of charge variants in therapeutic mAbs. The most widely employed techniques include:
- Ion-Exchange Chromatography (IEX) separates mAb variants based on their surface charge differences using either salt or pH gradients. [31] [32] Cation-exchange chromatography is particularly effective for resolving basic variants, including C-terminal lysine species. [31]
- Capillary Zone Electrophoresis (CZE) has demonstrated remarkable relevance for separating mAb charge variants using relatively simple conditions with dynamic neutral coatings. [12]
- Capillary Isoelectric Focusing (cIEF/iCIEF) provides high-resolution separation of charge variants based on their isoelectric points (pI) and has been successfully applied for biosimilarity assessment. [12] [33]
Table 2: Comparison of Major Analytical Techniques for Charge Variant Analysis
| Technique | Separation Principle | Resolution | Throughput | MS Compatibility | Key Applications |
|---|---|---|---|---|---|
| IEX-HPLC | Surface charge interaction | Moderate to High | Moderate | Limited with salt gradients, better with pH gradients [31] | Routine quality control, quantification of acidic/basic variants [32] |
| CZE-UV | Electrophoretic mobility in free solution | High | High | Good with specialized interfaces [12] | Biosimilarity assessment, detailed variant profiling [12] |
| cIEF/iCIEF | Isoelectric point (pI) | Very High | High | Challenging | High-resolution separation, pI determination [12] [33] |
| LC-MS Intact Mass | Mass-to-charge ratio | Limited for variants with same mass | Moderate | Excellent | Identification of mass changes associated with modifications [12] |
| CE-ESI-MS/MS | Peptide mapping after proteolysis | Very High for peptides | Low | Excellent | Detailed PTM identification and localization [12] |
Advanced Structural Characterization Methods
For comprehensive understanding of charge variant impact, advanced characterization techniques are employed to identify specific modifications:
- Mass spectrometry coupled with separation techniques enables precise identification of modifications responsible for charge differences. [12] [31] Online ESI-MS following pH gradient IEX separation has become increasingly popular for direct characterization. [31]
- Tryptic digestion followed by CE-ESI-MS/MS provides detailed characterization of peptide mixtures, enabling complete sequence coverage and consistent identification of PTMs including N-glycosylation and aspartic acid isomerization. [12]
- CZE-UV fraction collection with systematic enrichment allows isolation of specific charge variants for subsequent detailed analysis, connecting separation profiles with specific structural modifications. [12]
Application Notes: Strategic Approaches for Charge Variant Control
Risk-Based Control Strategy Implementation
Implementing a risk-based control strategy is essential for managing charge heterogeneity throughout product development and commercialization. [30] This approach involves:
- Early risk assessment to identify charge heterogeneity as a potential critical quality attribute (CQA) based on the molecule's susceptibility to modifications and their potential impact on safety and efficacy. [30]
- Process understanding to determine how various process parameters influence charge variant profiles, enabling definition of appropriate control strategies. [30]
- Phase-appropriate specifications that evolve through development stages, becoming more refined as product and process knowledge increases. [30]
- Comparability protocols to manage process changes while demonstrating consistent product quality. [30]
Machine Learning-Driven Optimization of Process Conditions
Machine learning (ML) approaches present a transformative opportunity for optimizing culture conditions to control charge variants. [18] Traditional methods like one-factor-at-a-time (OFAT) and design of experiments (DOE) often fail to capture the complex, nonlinear interactions between culture parameters and charge heterogeneity. [18] ML algorithms can:
- Analyze large datasets to uncover hidden patterns between process parameters (pH, temperature, duration) and charge variant profiles. [18]
- Predict optimal culture conditions for controlling specific charge variants, even when the underlying mechanisms are not fully understood. [18]
- Enable adaptive, ML-driven optimization strategies aligned with Quality-by-Design principles. [18]
Case studies have demonstrated ML's effectiveness in linking culture parameters to charge variants and providing insights for reducing acidic and basic variants. [18]
Experimental Protocols
Protocol 1: Charge Variant Analysis by Cation-Exchange HPLC
Purpose: To separate and quantify charge variants of monoclonal antibodies using weak cation-exchange chromatography (WCX-HPLC).
Materials and Equipment:
- Weak cation-exchange HPLC column (e.g., Thermo Scientific ProPac Elite WCX) [32]
- HPLC system with UV detection capability
- Mobile phase A: 10 mM sodium phosphate, pH 6.8
- Mobile phase B: 10 mM sodium phosphate, 500 mM sodium chloride, pH 6.8
- Sample preparation: Dilute mAb sample to 1 mg/mL in mobile phase A
Procedure:
- Equilibrate the WCX column with 95% mobile phase A and 5% mobile phase B for at least 30 minutes at flow rate 0.8 mL/min.
- Set column temperature to 30°C and UV detection to 280 nm.
- Inject 20 μL of prepared sample (1 mg/mL).
- Run a linear salt gradient from 5% to 45% mobile phase B over 45 minutes.
- Monitor elution profile and integrate peaks for acidic, main, and basic species.
- Regenerate column with 100% mobile phase B for 10 minutes before returning to initial conditions.
Data Analysis: Calculate relative percentages of acidic, main, and basic species based on peak area percentages. Compare with established reference standards or specifications.
Protocol 2: Charge Variant Characterization by CZE-UV with Fraction Collection
Purpose: To separate charge variants by capillary zone electrophoresis and collect fractions for subsequent structural characterization.
Materials and Equipment:
- Capillary electrophoresis system with UV detection and fraction collection capability
- Fused-silica capillary with dynamic neutral coating [12]
- Background electrolyte: 100 mM ε-aminocaproic acid with 0.05% hydroxypropylmethylcellulose, pH 5.0 [12]
- Sample preparation: Desalt mAb sample and dilute to 0.5 mg/mL in water
Procedure:
- Rinse capillary with background electrolyte for 5 minutes before each run.
- Inject sample hydrodynamically at 0.5 psi for 20 seconds.
- Apply separation voltage of 20 kV with the anode at the inlet.
- Monitor separation at 214 nm and collect fractions corresponding to individual charge variants.
- Concentrate collected fractions using centrifugal filters for subsequent analysis.
- For structural characterization, subject fractions to tryptic digestion followed by CE-ESI-MS/MS analysis. [12]
Data Analysis: Identify specific post-translational modifications in each fraction by comparing detected peptides with theoretical digest and noting mass shifts corresponding to known modifications.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Charge Variant Analysis
| Reagent/Material | Function | Application Notes | Key Suppliers |
|---|---|---|---|
| WCX HPLC Columns | Separation of basic variants and C-terminal lysine species | Optimal for mAbs with pI >6; compatible with pH or salt gradients [32] | Thermo Fisher, others |
| SCX HPLC Columns | Complementary selectivity to WCX | Enables faster gradients over wider pH range [32] | Thermo Fisher, others |
| SAX HPLC Columns | Separation of acidic variants | Optimal for proteins with pI <6 [32] | Thermo Fisher, others |
| Dynamic Coated Capillaries | Reduce electroosmotic flow and protein adsorption in CZE | Essential for robust CZE separations of mAbs [12] | Multiple CE suppliers |
| pH Gradient Buffers | Enable MS-compatible IEX separations | CX-1 buffer system allows linear pH gradients [32] | Thermo Fisher, others |
| IEF Markers | pI calibration for cIEF | Critical for accurate pI determination | Multiple suppliers |
| Trypsin | Proteolytic digestion for peptide mapping | Essential for detailed PTM identification by MS [12] | Multiple suppliers |
| Background Electrolytes | Separation medium for CZE | ε-Aminocaproic acid-based systems provide optimal separation [12] | Multiple suppliers |
Charge variant analysis remains an indispensable component of monoclonal antibody development and quality control. The critical link between charge heterogeneity and drug safety, efficacy, and stability necessitates comprehensive characterization throughout the product lifecycle. Implementation of robust analytical methods, coupled with strategic control approaches and emerging technologies like machine learning, enables manufacturers to ensure consistent product quality while mitigating risks associated with charge variants. As the biopharmaceutical landscape continues to evolve with increasing complexity of therapeutic modalities, the principles and methodologies outlined in these application notes provide a foundation for maintaining product quality and patient safety.
This application note details the regulatory requirements and practical analytical protocols for comprehensive characterization of charge variants in monoclonal antibody (mAb) therapeutics, as mandated by ICH Q6B guidelines. We provide detailed methodologies for charge variant separation, identification, and characterization, supported by case study data demonstrating the criticality of these analyses for biopharmaceutical development. The documented protocols enable researchers to establish robust control strategies ensuring product quality, consistency, and regulatory compliance.
The International Council for Harmonisation (ICH) Q6B guideline establishes a uniform set of international specifications for the structural characterization and quality control of biotechnological and biological products [34]. For monoclonal antibodies and related products, which exhibit inherent heterogeneity due to enzymatic and chemical modifications, ICH Q6B requires a thorough assessment of molecular properties to ensure product consistency, safety, and efficacy [34] [35].
Charge heterogeneity analysis is a fundamental requirement under ICH Q6B, as subtle variations in charge profiles can potentially impact biological activity, stability, and pharmacokinetics [36] [19]. These charge variants arise from post-translational modifications (PTMs) and chemical degradation occurring during manufacturing, purification, and storage [19] [37]. This document provides a detailed experimental framework for complying with ICH Q6B through comprehensive charge variant analysis.
Understanding Charge Variants in mAbs
Monoclonal antibodies are large, heterogeneous molecules subject to a variety of modifications that alter their surface charge distribution and isoelectric point (pI). The table below summarizes the major modifications leading to charge heterogeneity.
Table 1: Major Modifications Leading to Charge Variants in mAbs
| Variant Type | Modification | Chemical Basis | Effect on Charge |
|---|---|---|---|
| Acidic Species | Deamidation (Asn → Asp/isoAsp) [37] | Formation of aspartic acid or isoaspartic acid | Increases negative charge |
| Sialylation [37] | Addition of sialic acid residues | Increases negative charge | |
| C-terminal Lysine Cleavage [19] | Enzymatic removal of lysine | Reduces positive charge | |
| Glycation [19] | Non-enzymatic adduct formation with sugars | Can mask positive charges | |
| Disulfide Bond Reduction [37] | Breakage of S-S bonds | Can alter conformation and charge exposure | |
| Basic Species | C-terminal Amidation [19] | Addition of an amine group | Increases positive charge |
| Succinimide Formation [19] | Cyclization of aspartic acid | Eliminates a negative charge | |
| Oxidation (Met, Trp, His) [19] | Addition of oxygen to side chains | Can alter pKa of residues | |
| Incomplete N-terminal Pyroglutamate Formation [37] | Retention of uncyclized glutamine | Exposes a positive charge (NH₂) |
The following diagram illustrates the logical relationship between the sources of heterogeneity, the analytical techniques used for separation and characterization, and the final assessment required by regulators.
Analytical Protocols for Charge Variant Analysis
Charge Variant Separation by Cation-Exchange Chromatography (CEX)
This protocol describes the separation of mAb charge variants using a pH gradient on a strong cation-exchange (SCX) column, a high-resolution technique that offers advantages over traditional salt gradients and capillary isoelectric focusing (cIEF) in terms of robustness, reproducibility, and preparative scalability [36].
Materials and Reagents:
- Strong Cation-Exchange Column: e.g., ProPac SCX-10 (2 mm x 250 mm, 5 µm) [36]
- Mobile Phase A: 10 mM Sodium Phosphate Buffer, pH 6.0
- Mobile Phase B: 10 mM Sodium Phosphate Buffer, pH 9.5 + 250 mM NaCl (for salt gradient screening)
- pH Gradient Buffer Kit: Commercially available linear pH gradient buffer kits (e.g., Thermo Scientific) are recommended to ensure a truly linear pH gradient, which is critical for method robustness and reproducibility [36].
- mAb Sample: 1-2 mg/mL in a low-salt buffer (e.g., 10 mM Histidine, pH 6.0)
Instrumentation:
- UHPLC or HPLC system with binary or quaternary pump, autosampler, and UV-Vis detector
Method Parameters:
- Column Temperature: 30 °C
- Detection: UV at 280 nm
- Injection Volume: 10 µL
- Flow Rate: 0.2 mL/min
- Gradient Program (Linear pH Gradient):
- 0-5 min: 0% B (100% A, ~pH 6.0)
- 5-35 min: 0-100% B (linear gradient to ~pH 9.5)
- 35-40 min: 100% B
- 40-45 min: 0% B (re-equilibration)
Fraction Collection:
- Collect peaks corresponding to acidic variants, main species, and basic variants for further characterization [19]. Assess fraction purity by re-injecting an aliquot onto the same CEX method; purity should be >90% [19].
Structural Characterization of Isolated Variants
Once separated, isolated charge variants must be characterized to identify the specific chemical modifications, as required by ICH Q6B for product understanding and quality control [35].
A. Peptide Mapping for Primary Structure and Modifications
Procedure:
- Denaturation and Reduction: Dilute the isolated charge variant to 1 mg/mL in Guanidine HCl (6 M), Tris buffer (pH 8.0). Add Dithiothreitol (DTT) to 5 mM and incubate at 37 °C for 30 min.
- Alkylation: Add Iodoacetamide (IAA) to a final concentration of 10 mM. Incubate in the dark at room temperature for 30 min.
- Digestion: Desalt the protein into a digestion-compatible buffer (e.g., 50 mM Tris, pH 8.0). Add trypsin at a 1:20 (w/w) enzyme-to-substrate ratio. Incubate at 37 °C for 4-16 hours.
- LC-MS Analysis: Separate the resulting peptides using a C18 reversed-phase UHPLC column coupled to a high-resolution mass spectrometer (e.g., Q-TOF). Use a gradient of water/acetonitrile with 0.1% formic acid.
- Data Analysis: Use software to compare the measured peptide masses and MS/MS fragmentation spectra against the theoretical digest of the mAb sequence. Identify modified peptides (e.g., with deamidation, oxidation) by their mass shifts and confirm their identities with MS/MS [35].
B. Size Exclusion Chromatography (SEC) for Aggregation
Procedure:
- Column: SEC column (e.g., TSKgel G3000SWxl)
- Mobile Phase: 100 mM Sodium Phosphate, 100 mM Sodium Sulfate, pH 6.8
- Flow Rate: 0.5 mL/min
- Detection: UV at 280 nm
- Injection: 50 µg of isolated charge variant
- Analysis: Quantify the percentage of high-molecular-weight (HMW) aggregates and low-molecular-weight (LMW) fragments. Basic variants often show slightly elevated aggregate levels [19].
Case Study: Characterization of a Recombinant IgG1
The following data, representative of a typical characterization study, is derived from published work on a recombinant humanized IgG1 [19] [37].
Table 2: Analytical Characterization of Isolated Charge Variants from a Humanized IgG1
| Analytical Attribute | Starting Material | Acidic Variants | Main Species | Basic Variants | Technique Used |
|---|---|---|---|---|---|
| Proportion | 100% | 20% | 68% | 12% | CEX Analysis [19] |
| Purity (by CEX) | N/A | 95% | 94% | 94% | CEX Re-injection [19] |
| Aggregates (by SEC) | <0.3% | <0.3% | <0.3% | ~10% | Size Exclusion Chromatography [19] |
| Notable Modifications | Mixture | Deamidation (CH2/CH3), Sialylation [37] | N-terminal pyroGlu, No C-terminal Lys [37] | C-terminal Lys, Succinimide, Oxidation [19] [37] | Peptide Mapping & LC-MS |
| In Vitro Potency | Reference | Comparable | Comparable | Comparable | Cell-based Assay [19] |
| FcRn Binding | Reference | Comparable | Comparable | Comparable | Surface Plasmon Resonance [19] |
The Scientist's Toolkit: Essential Reagents and Materials
The following table lists key materials required for the charge variant analysis workflows described in this note.
Table 3: Key Research Reagent Solutions for Charge Variant Analysis
| Item | Function/Application | Example/Best Practice |
|---|---|---|
| Cation-Exchange Column | High-resolution separation of charge variants based on surface charge differences. | ProPac SCX-10 column; use of small particle sizes (<5 µm) for UHPLC application is recommended [36]. |
| pH Gradient Buffer Kits | Provide matched buffer pairs for generating robust, reproducible, and linear pH gradients in CEX. | Commercially available kits overcome the challenge of creating truly linear gradients with homemade buffer cocktails [36]. |
| Trypsin, Sequencing Grade | Proteolytic enzyme for digesting mAbs into peptides for peptide mapping. | High-purity, sequencing-grade enzyme minimizes autolysis and ensures reproducible digestion [35]. |
| UHPLC-MS System | High-resolution separation (UHPLC) and accurate mass detection (MS) for peptide mapping and intact mass analysis. | Q-TOF instruments provide high mass accuracy for confident identification of modifications [35]. |
| Reference Standard | Well-characterized mAb material used for system suitability testing and method qualification. | NISTmAb is a widely used reference standard for benchmarking charge variant methods [36]. |
Compliance with ICH Q6B requires a systematic and multi-faceted approach to characterizing therapeutic proteins. As demonstrated, charge variant analysis is a critical component of the control strategy for monoclonal antibodies. The application of robust, pH-gradient CEX methods, coupled with detailed characterization techniques like peptide mapping, provides the necessary data to understand product heterogeneity. While charge variants are often present, rigorous assessment as outlined in this note can demonstrate that these variants, when controlled within defined limits, have no detrimental impact on product quality, potency, or pharmacokinetics, thereby ensuring the safety and efficacy of the biotherapeutic product [19].
Analytical Toolkit: Methodologies for Separating and Characterizing Charge Variants
In the characterization of monoclonal antibodies (mAbs) and other biotherapeutics, charge variant analysis constitutes a critical quality assessment mandated by regulatory authorities [36]. Ion-exchange chromatography (IEC) is a pivotal technique for separating these charge variants, primarily operating through two elution modes: salt gradients and pH gradients [38]. The choice between these elution methods significantly impacts selectivity, resolution, method development efficiency, and compatibility with downstream detection systems like mass spectrometry (MS) [28] [31]. This application note provides a detailed comparison of salt- and pH-based gradient elution for the analysis of monoclonal antibody charge variants. It is structured within the broader research context of ensuring drug efficacy, stability, and safety by monitoring critical quality attributes such as lysine variants, deamidation, sialylation, and oxidation [31].
Comparative Analysis: Salt vs. pH Gradients
The fundamental mechanism of IEC involves the electrostatic interaction between charged functional groups on a protein and the oppositely charged ligands on the chromatographic stationary phase. Positively charged mAbs (at a mobile phase pH below their isoelectric point (pI)) are typically separated by Cation Exchange Chromatography (CEX), while Anion Exchange Chromatography (AEX) is used for negatively charged impurities or specific analyte classes [39]. Elution is achieved by disrupting these interactions, either by increasing the ionic strength with salt (e.g., NaCl) or by altering the net charge of the protein through a pH shift [36] [39].
The table below summarizes the core characteristics of the two primary elution methods.
Table 1: Comparative analysis of salt gradient versus pH gradient elution in IEC
| Parameter | Salt Gradient Elution | pH Gradient Elution |
|---|---|---|
| Elution Principle | Competitive displacement using increasing concentration of counter-ions (e.g., NaCl, KCl) [40]. | Modifying the net surface charge of the analyte until it no longer binds to the stationary phase [36]. |
| Selectivity & Resolution | High selectivity for variants with subtle charge differences; resolution is highly dependent on gradient slope and salt type [40]. | Provides high selectivity and can separate variants that may co-elute under salt gradients; offers a different selectivity profile [36] [31]. |
| Method Development | Often requires extensive, molecule-specific optimization of gradient slope and starting conditions, making it time-consuming [36]. | More generic and predictable; a single, standardized method can often be applied to a wide range of mAbs, simplifying development [36] [27]. |
| MS-Compatibility | Traditionally poor due to non-volatile salts in the mobile phase, requiring extensive desalting or buffer exchange prior to MS [28] [31]. | Inherently more compatible as volatile ammonium-based buffers (e.g., ammonium acetate/formate) can be used, enabling direct online coupling [28] [31]. |
| Typical Run Time | Can be long; often up to 90 minutes for high-resolution separations [36]. | Generally faster; run times of 30 minutes or less are achievable with UHPLC systems [36]. |
| Reproducibility & Robustness | Can suffer from poor reproducibility due to challenges in precisely controlling salt gradients and buffer preparation [36]. | High reproducibility, especially when using commercially available pre-mixed pH gradient buffer kits that ensure linear and consistent gradients [36] [27]. |
| Primary Application | Historically the most common approach; widely used in both analytical and process-scale purification [40] [39]. | Gaining prominence for high-throughput, robust analytical characterization and quality control of mAbs [36] [38]. |
Experimental Protocols
Protocol A: CEX with Linear Salt Gradient
This protocol describes the separation of mAb charge variants using a linear salt gradient on a cation-exchange column, suitable for both analytical characterization and preparative purification [40] [39].
Materials:
- Column: Thermo Scientific MAbPac SCX-10 column (3 µm particle size, 4.6 x 50 mm) or equivalent [27].
- Mobile Phase A: 50 mM Sodium phosphate, pH 6.0.
- Mobile Phase B: 50 mM Sodium phosphate, 500 mM Sodium Chloride (NaCl), pH 6.0.
- Sample: Monoclonal antibody, 1 mg/mL in Mobile Phase A.
- System: UHPLC system with UV detection (e.g., Thermo Scientific Vanquish Flex).
Method:
- System Equilibration: Equilibrate the column with 5-10 column volumes (CV) of 10% Mobile Phase B (equivalent to 50 mM NaCl) at a flow rate of 0.5 mL/min. Monitor the UV baseline at 280 nm until stable.
- Sample Injection: Inject 10 µL of the prepared mAb sample.
- Gradient Elution: Immediately initiate a linear gradient from 10% B to 60% B over 30 minutes. This corresponds to an increase from 50 mM to 300 mM NaCl.
- Column Cleaning & Re-equilibration: After the gradient, flush the column with 100% B for 5 CV to remove strongly bound impurities. Re-equilibrate with 10% B for at least 10 CV before the next run.
- Data Analysis: Identify the main mAb peak, along with acidic variants (eluting earlier) and basic variants (eluting later) [31]. Integrate peak areas for quantification.
Protocol B: CEX with Linear pH Gradient
This protocol utilizes a pH gradient for the generic and robust separation of mAb charge variants, optimized for coupling with mass spectrometry.
Materials:
- Column: Thermo Scientific ProPac WCX-10 column (5 µm particle size, 4 x 250 mm) [27].
- Buffers: Commercial pH gradient buffer kit (e.g., Thermo Scientific pH Gradient Buffer Kit), with Buffer A (low pH, e.g., pH 5.6) and Buffer B (high pH, e.g., pH 10.0) [36] [27].
- Mobile Phase A: pH Gradient Buffer A.
- Mobile Phase B: pH Gradient Buffer B.
- Sample: Monoclonal antibody, 2 mg/mL in water or a mild buffer.
- System: UHPLC system coupled to a mass spectrometer.
Method:
- System Equilibration: Equilibrate the column with 100% Mobile Phase A for at least 10 CV at a flow rate of 0.8 mL/min.
- Sample Injection: Inject 5 µL of the mAb sample.
- Gradient Elution: Initiate a linear gradient from 0% B to 100% B over 25 column volumes.
- MS Coupling: Directly couple the column outlet to the ESI-MS source. Use MS-compatible conditions (e.g., nitrogen gas, appropriate vaporizer temperature) and acquire data in the m/z range of 2000-4000 for intact protein analysis [28].
- Data Analysis: Correlate the UV chromatogram with the extracted ion chromatograms (XICs) and deconvoluted masses from the MS data to identify specific proteoforms like lysine truncations or glycosylation variants [28].
Decision Workflow for Method Selection
The following diagram illustrates a logical pathway for selecting the appropriate IEC elution method based on project goals and constraints.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of IEC methods relies on a set of core materials. The following table lists key reagent solutions and their functions.
Table 2: Essential materials and reagents for IEC charge variant analysis
| Item | Function & Importance |
|---|---|
| Cation-Exchange Column (e.g., ProPac WCX-10, MAbPac SCX-10) [27] | The stationary phase that separates charge variants based on electrostatic interactions. A high-resolution, hydrophilic column is crucial for resolving subtle variants. |
| pH Gradient Buffer Kits [36] [27] | Pre-mixed, MS-compatible buffers designed to produce a linear and reproducible pH gradient, saving method development time and ensuring robustness. |
| Volatile Salts (Ammonium acetate, Ammonium formate) [28] | MS-compatible salts used for creating salt gradients or as additives in pH gradient methods when online MS detection is employed. |
| Non-Volatile Salts (Sodium chloride, Sodium phosphate) [40] | Traditional salts for preparative-scale purification or analytical methods where MS coupling is not required. |
| UHPLC System (e.g., Thermo Scientific Vanquish Flex) [27] | Provides high-pressure capability, precise gradient formation, and low-dispersion fluidics for fast, high-resolution separations. |
| Mass Spectrometer (Q-TOF, Orbitrap) [28] [38] | Used for the definitive identification of charge variants (e.g., lysine variants, glycosylation, deamidation) by intact mass analysis or after fraction collection. |
Both salt and pH gradient elution methods are powerful tools for charge variant analysis of monoclonal antibodies. The optimal choice is dictated by the specific application. Salt gradients remain the workhorse for process-scale purification and can offer high resolution for specific, well-characterized molecules. In contrast, pH gradients provide a more generic, robust, and MS-friendly platform for analytical characterization and quality control, significantly accelerating method development and transfer. A mechanistic understanding of both approaches, as facilitated by this application note, empowers scientists to make informed decisions that enhance the efficiency and effectiveness of biopharmaceutical development.
Charge heterogeneity is a Critical Quality Attribute (CQA) for monoclonal antibody (mAb) therapeutics, as it can impact product safety, efficacy, and stability. Post-translational modifications such as deamidation, oxidation, glycosylation, and C-terminal lysine clipping introduce charge variants that must be carefully monitored and controlled throughout the biopharmaceutical development lifecycle [41] [12]. Imaged capillary isoelectric focusing (icIEF) has emerged as the industry's gold-standard technique for high-resolution charge variant analysis, leveraging pH gradients to separate mAb variants based on their isoelectric point (pI) with exceptional resolution, reproducibility, and regulatory compliance [42] [41].
This Application Note details optimized icIEF methodologies for the characterization of mAb charge heterogeneity, providing researchers and drug development professionals with validated protocols for implementation in quality control environments. The procedures outlined herein enable precise identity testing and purity assessment of biotherapeutic products, supporting comparability studies and biosimilarity assessments [12].
Key Research Reagent Solutions
The following reagents and materials are essential for implementing robust icIEF methodologies for mAb charge variant analysis.
Table 1: Essential Reagents and Materials for icIEF Analysis
| Reagent/Material | Function/Purpose | Application Notes |
|---|---|---|
| Carrier Ampholytes (CAs) | Generate pH gradient for separation | Critical for establishing stable, linear pH gradient; concentration typically 2-4% [41] |
| Methylcellulose | Acts as sieving matrix and stabilizer | Reduces electroosmotic flow (EOF) and analyte-wall interactions; concentration 0.1-1% [41] |
| pI Markers (pIMs) | Internal standards for pI calibration | Essential for assigning pI values to sample peaks; use chemically defined markers [41] |
| Urea | Denaturant for sample preparation | Improves solubility and prevents aggregation; typically 4-6 M concentration [41] |
| L-Arginine (L-Arg) | Additive for solubility enhancement | Reduces protein aggregation and improves peak shape; commonly 25-100 mM [41] |
| Pharmalyte | Specific type of carrier ampholyte | Creates precise pH gradients (e.g., pH 3-10, 5-8) for optimal mAb separation [41] |
icIEF Experimental Protocol
Sample Preparation
Desalting and Buffer Exchange: Transfer mAb samples into a low-conductivity solution such as 10 mM sodium chloride or deionized water using centrifugal filtration devices (10 kDa MWCO). Final mAb concentration should be 0.5-2 mg/mL [41].
Master Mix Preparation: Prepare the icIEF master mix according to the following formulation:
- 4% carrier ampholytes (pH 5-8 for mAbs)
- 0.35% methylcellulose
- 2 M urea
- 50 mM L-arginine
- pI markers: 0.1-0.2% each appropriate for the expected pI range [41]
Sample Mix Preparation: Combine desalted mAb sample with master mix at a 4:1 ratio (v/v) to achieve final mAb concentration of 0.1-0.4 mg/mL. Centrifuge at 10,000 × g for 5 minutes to remove particulates [41].
Instrument Setup and Separation
Capillary Selection: Use fluorocarbon- or acrylamide-coated capillaries (50-100 μm ID, 5-10 cm length) to suppress electroosmotic flow and minimize protein adsorption [41].
Instrument Parameters:
- Pre-focusing: 1.0 kV for 1 minute
- Focusing: 3.0 kV for 8-10 minutes
- Total analysis time: Typically <10 minutes
- Detection: UV at 280 nm [41]
Anolyte/Catholyte Preparation:
- Anolyte: 80 mM phosphoric acid
- Catholyte: 100 mM sodium hydroxide [41]
Data Analysis
pI Determination: Calculate pI values of sample peaks by linear regression using known pI markers.
Variant Quantification: Integrate peak areas for acidic, main, and basic variants. Express each variant as a percentage of total peak area.
System Suitability: Verify resolution between critical peak pairs (typically ≥2.0) and pI marker linearity (R² ≥ 0.99) [41].
icIEF Mechanism and Workflow
The following diagram illustrates the fundamental separation mechanism and procedural workflow of icIEF analysis.
Method Validation and Performance Data
The icIEF method demonstrates excellent performance characteristics suitable for regulatory submission and quality control. The following table summarizes typical validation results obtained for mAb charge variant analysis.
Table 2: icIEF Method Validation Data for mAb Charge Variant Analysis
| Performance Parameter | Acceptance Criteria | Typical Results | Regulatory Reference |
|---|---|---|---|
| Precision (Peak Area %RSD) | ≤ 10% | ≤ 5% | ICH Q2(R2) [42] |
| Precision (pI %RSD) | ≤ 1% | ≤ 0.5% | ICH Q2(R2) [42] |
| Linearity (R²) | ≥ 0.990 | ≥ 0.995 | ICH Q2(R2) [42] |
| Resolution | ≥ 2.0 | 2.5 - 4.0 | Ph. Eur. [42] [41] |
| Accuracy/Recovery | 90-110% | 95-105% | ICH Q2(R2) [42] |
| Range | 50-150% of target | 25-200% of target | ICH Q2(R2) [42] |
Application to Biosimilarity Assessment
icIEF provides a powerful tool for biosimilarity assessment, as demonstrated in the analysis of infliximab innovator and biosimilar products. When applied to intact mAbs, icIEF can distinguish between products with subtle differences in charge variant profiles [12]. The technique has been successfully implemented for:
- Identity testing based on unique charge heterogeneity profiles
- Batch-to-batch consistency monitoring
- Stability studies tracking charge variant formation over time
- Biosimilarity demonstration through comparative profile analysis [41] [12]
Troubleshooting Guide
Table 3: Common icIEF Issues and Solutions
| Problem | Potential Causes | Recommended Solutions |
|---|---|---|
| Poor resolution | Inadequate ampholyte blend, insufficient focusing time | Optimize pH range of ampholytes, increase focusing time or voltage [41] |
| Peak tailing | Protein adsorption to capillary, aggregation | Ensure proper capillary coating, add urea or L-arginine to sample [41] |
| Irreproducible migration | Electroosmotic flow, unstable gradient | Use appropriate capillary coating, fresh ampholytes, check chemical stability [41] |
| Broad peaks | Overloading, incorrect focusing parameters | Reduce sample concentration, optimize focusing time and voltage [41] |
icIEF represents a robust, high-resolution platform for charge variant analysis of monoclonal antibodies and other biotherapeutics. The methodology outlined in this Application Note provides researchers with a validated approach for characterizing charge heterogeneity, supporting product development, quality control, and regulatory submissions. With its exceptional reproducibility, sensitivity, and compliance with pharmacopeial standards, icIEF continues to be the industry's gold-standard technique for ensuring the safety and efficacy of biopharmaceutical products.
Within the development of monoclonal antibody (mAb) therapeutics, charge variant analysis stands as a critical quality attribute required by regulatory authorities for ensuring the safety, efficacy, and consistency of biologic drugs [43]. Charge heterogeneity, comprising acidic, main, and basic species, arises from post-translational modifications such as deamidation, oxidation, C-terminal lysine clipping, and glycosylation variations that occur during production and storage [44] [43]. For biosimilar developers, even minor differences in charge variant profiles compared to the innovator product necessitate comprehensive characterization, as these can impact biological functions like antigen and receptor binding [44].
Fraction collection is the strategic physical separation of these charge variants, typically via chromatographic or electrophoretic methods, to enable detailed structural and functional interrogation of isolated species. This application note details robust protocols for the fractionation of mAb charge variants using cation-exchange chromatography (CEX), preparing purified acidic, main, and basic species for downstream mass spectrometric and functional analyses.
Materials and Methods
Research Reagent Solutions
The following table catalogues essential materials and reagents for the fractionation process and subsequent analysis.
Table 1: Key Research Reagents and Materials
| Item | Function/Application | Reference |
|---|---|---|
| BioPro SPF Non-porous Column (5 µm, 100 × 4.6 mm) | Stationary phase for analytical and preparative CEX separation of charge variants. | [44] |
| Ammonium Acetate (MS-grade) | MS-compatible volatile salt for mobile phase preparation in CEX-MS coupling. | [28] [45] |
| Sodium Phosphate Buffer | Non-volatile salt-based mobile phase for high-resolution CEX-UV fractionation. | [44] |
| ACQUITY UPLC-BEH300 C4 Column (1.7 µm) | Stationary phase for reversed-phase LC-MS intact mass analysis of fractions. | [44] |
| Xevo G2-XS QToF Mass Spectrometer | High-resolution mass spectrometer for accurate mass determination of intact proteins and peptides. | [44] |
| Immunoglobulin G-degrading enzyme (IdeS) | Protease for digesting mAbs into Fc and F(ab')2 subunits for localized variant analysis. | [45] |
| PNGase F (Peptide:N-glycosidase F) | Enzyme for removing N-linked glycans, converting Asn to Asp and lowering pI for improved separation. | [45] |
CEX-Based Fractionation Workflow
The overarching process for preparing charge variants for downstream analysis, from initial separation to functional characterization, is illustrated below.
Protocol 1: Preparative CEX Fractionation for Downstream Analysis
This protocol is adapted from the method used for the detailed characterization of Avastin and its biosimilar candidate, SIMAB054 [44]. It is designed for high-concentration loading to yield sufficient fractions.
Equipment & Materials
- HPLC system with PDA detector and automated fraction collector (e.g., Waters ACQUITY)
- BioPro SPF Non-porous Column (5 µm, 100 × 4.6 mm, YMC)
- Mobile Phase A: 100 mM Sodium Dihydrogen Phosphate Dihydrate (NaH₂PO₄·2H₂O) in MS-grade water.
- Mobile Phase B: 100 mM Disodium Hydrogen Phosphate Dihydrate (Na₂HPO₄·2H₂O) in MS-grade water.
- Mobile Phase C: 1 M Sodium Chloride (NaCl) in MS-grade water.
- mAb sample (e.g., diluted to 9.4 mg/mL with ultrapure water).
Chromatographic Procedure
- System Preparation: Equilibrate the column with the starting mobile phase (a blend of A and B to achieve pH 5.7) at a flow rate of 0.5 mL/min. Column temperature should be maintained at 25°C.
- Sample Injection: Inject the high-concentration mAb sample (e.g., 9.4 mg/mL) directly onto the column.
- Gradient Elution: Execute a salt gradient from 0 mM to 200 mM sodium phosphate at a constant pH of 5.7 using the system's "Auto Blend" function. This method precisely controls the mixture of Mobile Phases A and B to maintain the pH while increasing ionic strength.
- UV Detection & Fraction Collection: Monitor the eluent at 280 nm and 214 nm. Using the automated fraction manager, collect the distinct peaks corresponding to the acidic species (first major peak(s)), the main species (central major peak), and the basic species (last major peak(s)) into separate vials.
Protocol 2: A Generic CEX-MS Method for Intact Analysis
This protocol provides a generic, MS-compatible approach for online characterization of charge variants across multiple mAbs, as reviewed in recent literature [28]. It uses volatile ammonium acetate buffers.
Equipment & Materials
- LC-MS system equipped with nanoESI source and online UV detector.
- Cation-exchange column (e.g., BioPro IEX SF).
- Eluent A: 50 mM Ammonium Acetate + 2% Acetonitrile, pH 5.0.
- Eluent B: 100 mM Ammonium Acetate + 2% Acetonitrile, pH 8.5.
- mAb sample (2 mg/mL).
Chromatographic Procedure
- System Setup: Equilibrate the column with 55% Eluent B at a reduced flow rate of 0.1 mL/min to optimize MS compatibility and separation.
- Sample Injection: Inject the mAb sample.
- Gradient Elution:
- Hold at 55% B for 2 minutes.
- Apply a linear gradient from 55% B to 85% B over 25 minutes.
- This prolonged, shallow gradient is key to achieving baseline separation of acidic and basic variants.
- Detection: Monitor separation via online UV (280 nm). The eluent is then split, with a sub-µL/min flow directed to the nanoESI-MS, which should be operated in positive ion mode with a high m/z range (e.g., 4500-6500) to decluster ammonia adducts and detect the high-mass ions [28].
Results and Data Interpretation
Structural Analysis of Collected Fractions
Following fractionation, the collected variants are subjected to detailed structural analysis to identify the modifications responsible for charge differences.
- Intact Mass Analysis: As performed in the Avastin biosimilar study, fractions are buffer-exchanged and analyzed by UPLC-ESI-QToF under denaturing conditions (e.g., C4 column with water/ACN/0.1% FA gradient). This reveals mass differences associated with C-terminal lysine clipping (∼+128 Da per lysine), glycoforms (e.g., afucosylation, galactosylation), and oxidation [44].
- Peptide Mapping: Fractions are denatured, reduced, alkylated, and digested with trypsin. The resulting peptides are analyzed by RP-LC-MS/MS. This identifies site-specific modifications like deamidation of asparagine (+1 Da), methionine oxidation (+16 Da), and N-terminal pyroglutamic acid formation (-17 Da) [44] [43].
- Subunit Analysis: Treatment of fractions with IdeS protease cleaves mAbs into Fc and F(ab')2 fragments. Subsequent LC-MS analysis of these subunits localizes charge variants to specific domains, which is critical for functional assessment [45].
Functional Analysis of Charge Variants
The biological impact of charge modifications is assessed by testing the binding affinity of isolated fractions.
- Surface Plasmon Resonance (SPR): This is the gold-standard for in vitro functional analysis. The binding affinity (KD) of the acidic, main, and basic fractions against the target antigen (e.g., VEGF-A for Avastin) and the neonatal Fc receptor (FcRn) is determined. Studies have shown that variants, particularly those with modifications in the CDR or Fc region, can exhibit significantly different binding capacities, a key finding in a comparability exercise [44] [43].
Table 2: Key Structural and Functional Differences in Fractionated Charge Variants
| Charge Variant | Common Structural Attributes | Potential Functional Impact |
|---|---|---|
| Acidic Species | Deamidation (Asn), Sialylation, Glycation, N-terminal Pyro-Glu, Trisulfide bonds | May reduce antigen-binding affinity if deamidation occurs in CDR regions [44] [43]. Increased galactosylation has been observed [44]. |
| Main Species | Represents the desired product profile with minimal modifications. | Serves as the reference standard for binding affinity and biological activity. |
| Basic Species | C-terminal Lysine, C-terminal Amidation, N-terminal Glutamine, Low Glycosylation/Afucosylation | Unprocessed C-terminal lysine, which is clinically irrelevant as it is degraded in serum. However, afucosylation can enhance ADCC activity [44] [43]. |
Discussion
Strategic Considerations for Fraction Collection
The choice of fractionation strategy has significant implications for downstream analysis. The use of MS-compatible volatile buffers (e.g., ammonium acetate) in CEX-MS allows for direct online identification but may require careful method optimization to achieve resolution comparable to non-volatile systems [28]. In contrast, preparative fractionation with non-volatile salts (e.g., sodium phosphate), as detailed in Protocol 1, often provides superior peak resolution and higher protein recovery, which is crucial for subsequent offline structural and functional assays [44]. A key consideration is that fraction-based methods enhance sensitivity by eliminating interfering proteoforms, allowing for the precise determination of low-abundance modifications [43].
Technical Notes and Troubleshooting
- Resolution Optimization: For CEX methods, adjusting the initial %B (strong buffer) can dramatically impact the separation of acidic and basic variants from the main peak. A higher starting ionic strength can resolve variants but may cause peak broadening if too high [28].
- MS Coupling Challenges: Nano-electrospray ionization (nanoESI) is recommended for CEX-MS and AEX-MS due to its tolerance for higher salt concentrations (up to 600 mM ammonium acetate). Harsh declustering parameters in the MS source are often necessary to dissociate ammonia adducts [28] [45].
- Method Genericity: A single, generic CEX method can be applied to a panel of mAbs with varying pIs (e.g., from 7.3 to 8.7), but individual optimization for each mAb can significantly improve separation [28].
- Alternative Techniques: For mAbs with lower pIs (e.g., IgG4-based mAbs), Anion Exchange Chromatography (AEX) can be a more suitable separation technique than CEX, providing excellent separation of charge variants, including site-specific deamidations and glycosylation heterogeneity [45].
In the development and quality control of monoclonal antibody (mAb) therapeutics, charge variant analysis is a critical exercise for ensuring product efficacy, safety, and consistency. These charge variants often arise from a complex array of post-translational modifications (PTMs), which can alter the antibody's isoelectric point (pI), stability, and biological function [46] [47]. Mass spectrometry (MS) has emerged as an indispensable technology for characterizing these modifications, primarily through two complementary approaches: intact mass analysis and peptide mapping. This application note details robust protocols for both techniques, framing them within the essential context of mAb charge variant characterization for researchers and drug development professionals.
Intact Mass Analysis for High-Level Characterization
Intact mass analysis involves measuring the mass of the whole protein or large subunits with high accuracy, providing a global overview of the proteoform population, including major PTMs and modifications introduced during production [48].
Experimental Protocol
- Sample Preparation: For monoclonal antibodies, begin with >90% pure protein at a minimum concentration of 0.1-0.5 mg/mL. A total of 50 μg per analysis is typically required [48].
- Reduction (Optional): To study light and heavy chains separately, incubate the antibody with dithiothreitol (DDT) [48].
- Deglycosylation (Recommended): To simplify the mass spectrum by removing heterogeneous glycosylation, treat the antibody with PNGase F. Note that this converts asparagine to aspartic acid, resulting in a +1 Da mass shift per glycosylation site [48] [13].
- Enzymatic Digestion (Optional - Middle-Down): For a more detailed view of large domains, use enzymes like IdeS to generate Fab and Fc fragments [48].
- Liquid Chromatography (LC): Employ reversed-phase liquid chromatography (RPLC) using a wide-pore C4 or C8 column (e.g., 300 Å pore size) for protein separation. A water/acetonitrile gradient with 0.1% formic acid is standard.
- Mass Spectrometry (MS): Use high-resolution mass spectrometry, such as Q-TOF or Orbitrap instruments, coupled with electrospray ionization (ESI). ESI typically generates a charge envelope that must be deconvoluted to obtain the neutral intact mass [48].
- Data Analysis: Deconvolute the raw mass spectrum using software (e.g., MaxEnt, BioPharma Finder) to generate a zero-charge mass spectrum. Compare the observed mass(es) with the theoretical mass calculated from the amino acid sequence to identify mass shifts indicative of PTMs like lysine truncation, glycosylation, or oxidation [48].
Application in Charge Variant Analysis
Intact mass analysis, especially when performed on reduced chains or IdeS fragments, can directly link mass shifts to specific charge variants isolated by techniques like cation-exchange chromatography (CEX). For instance, the loss of 128 Da from the heavy chain can be confidently assigned to C-terminal lysine processing, a common source of charge heterogeneity [13] [47].
Figure 1: Experimental workflow for intact mass analysis of monoclonal antibodies.
Peptide Mapping for Residue-Level PTM Identification
While intact mass analysis identifies mass shifts, peptide mapping (a bottom-up approach) is required to pinpoint the exact location and structure of PTMs. This method provides residue-level confirmation of the amino acid sequence and its modifications [13] [49].
Experimental Protocol
- Denaturation, Reduction, and Alkylation: Dilute the antibody to 1-2 mg/mL. Denature with guanidine hydrochloride or urea, reduce with DTT, and alkylate cysteine residues with iodoacetamide.
- Enzymatic Digestion: Use a sequence-grade protease, most commonly trypsin, at an enzyme-to-substrate ratio of 1:20 to 1:50 (w/w). Incubate at 37°C for 4-18 hours. Alternative enzymes (e.g., Lys-C, Asp-N) can provide complementary coverage.
- Liquid Chromatography: Perform reversed-phase LC on a C18 column with a shallow water/acetonitrile gradient (typically 0.1% formic acid) to separate the complex peptide mixture.
- Tandem Mass Spectrometry (LC-MS/MS): Analyze the eluting peptides using a high-resolution tandem mass spectrometer. Data-Dependent Acquisition (DDA) is standard: a full MS1 scan is followed by MS/MS fragmentation of the most intense precursor ions.
- Data Analysis: Search the resulting MS/MS spectra against the expected protein sequence(s) using software (e.g., BioPharma Finder, Byos, Peaks). Use a narrow mass tolerance for precursors and fragments. Critical Step: Manually verify software assignments, particularly for isobaric or low-abundance modifications, to avoid misidentifications [49].
Application in Charge Variant Analysis
Peptide mapping is unparalleled for characterizing the molecular basis of charge variants separated by CEX. It can:
- Localize Deamidation: Identify the specific asparagine or glutamine residue responsible for the acidic shift.
- Identify Succinimide Intermediates: Detect the cyclic intermediate of deamidation.
- Quantify Oxidation: Measure the relative abundance of methionine or tryptophan oxidation.
- Confirm Sequence Variants: Distinguish true amino acid substitutions from isobaric PTMs [49].
Figure 2: Detailed workflow for peptide mapping to achieve residue-level PTM identification.
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential reagents and materials for mass spectrometry-based PTM analysis.
| Item | Function & Application | Key Considerations |
|---|---|---|
| PNGase F | Enzyme for removing N-linked glycans. Simplifies intact mass spectra and confirms glycosylation sites in peptide mapping. | Converts Asn to Asp (+1 Da mass shift); essential for confirming N-linked sites [13]. |
| IdeS / FabRICATOR | Protease that cleaves IgG below the hinge region, generating Fc and F(ab')2 fragments. Enables "middle-down" analysis. | Ideal for domain-specific characterization of PTMs and charge variants [48]. |
| Trypsin (Sequencing Grade) | Gold-standard protease for peptide mapping. Cleaves C-terminal to Lys and Arg, generating peptides ideal for MS/MS. | Must be sequencing grade to avoid autolysis; other enzymes (Lys-C) can be used in combination [49]. |
| Dithiothreitol (DTT) / Tris(2-carboxyethyl)phosphine (TCEP) | Reducing agents to break inter- and intra-chain disulfide bonds. | TCEP is more stable and does not require alkylation, but DTT is more common. |
| Iodoacetamide | Alkylating agent that caps reduced cysteine residues to prevent reformation of disulfide bonds. | Must be used in the dark; excess reagent must be quenched. |
| C4 / C18 LC Columns | C4 for intact protein/domain separation; C18 for peptide separation in mapping. | Wide-pore (300Å+) columns are necessary for large proteins and fragments. |
| Cation-Exchange (CEX) Columns | For initial separation of mAb charge variants (e.g., BioResolve SCX mAb Column). | pH-gradient elution has proven superior for separating mAbs with similar pIs [46] [47]. |
Data Presentation and Analysis
Software and Data Quality Considerations
The complexity of MS data from PTM analysis demands robust software and rigorous quality control. Commercial software packages are often used in regulated environments due to validation requirements, but they have limitations. A 2025 study highlighted that software can misidentify peptides due to isobaric dipeptides (e.g., LeuGlu vs. AsnLys, which have identical mass) and can introduce artifacts, such as assigning false succinylation to compensate for sequence mismatches [49]. Therefore, manual validation of MS/MS spectra is a non-negotiable step for unambiguous PTM localization and sequence confirmation.
For native mass spectrometry of intact complexes, new algorithms like precisION use a fragment-level open search to discover "hidden" modifications without prior knowledge, greatly enhancing the ability to characterize heterogeneous samples [50]. Adherence to community-developed data quality metrics is also crucial for ensuring the reproducibility and reliability of proteomic data, particularly in biomarker discovery and therapeutic development [51] [52].
Quantitative Data from PTM Analysis
Table 2: Common PTMs identified in monoclonal antibodies and their analytical signatures.
| PTM | Mass Shift (Δ, Da) | Effect on Charge | Primary Analytical Technique | Notes |
|---|---|---|---|---|
| C-terminal Lysine | -128.09 (loss) | More acidic | Intact Mass (Heavy Chain), Peptide Mapping | Common source of basic variants; fully processed form is acidic [47]. |
| Deamidation (Asn→Asp/isoAsp) | +0.984 | More acidic | Peptide Mapping | Can be resolved chromatographically; major driver of acidic variants [13]. |
| Methionine Oxidation | +15.995 | Slightly more acidic | Intact Mass, Peptide Mapping | Can impact stability and efficacy; monitored in stress studies. |
| N-terminal Pyroglutamate | -17.027 (from Gln) | More basic | Peptide Mapping, Intact Mass | Common on heavy and light chains; nearly 100% occupancy in some mAbs [49]. |
| Glycosylation (G0F) | +1444.527 (G0F) | Neutral / Complex | Intact Mass, Peptide Mapping (HILIC) | Critical quality attribute; requires HILIC or deglycosylation for detailed analysis [48] [13]. |
In the development and quality control of monoclonal antibodies (mAbs) and other biotherapeutics, comprehensive characterization of critical quality attributes (CQAs) is paramount. Among these CQAs, size-based variants (aggregates and fragments) and purity are essential indicators of product safety and efficacy. Charge variant analysis provides crucial information about the heterogeneity of mAbs, which can arise from post-translational modifications such as deamidation, C-terminal lysine clipping, and glycosylation variants [5]. This application note details the implementation of two orthogonal techniques: Size Exclusion Chromatography (SEC) for the analysis of aggregates and Capillary Electrophoresis-Sodium Dodecyl Sulfate (CE-SDS) for purity assessment, framed within the broader context of mAb charge variant characterization.
The complementary nature of these methods provides a comprehensive analytical profile. SEC separates molecules based on their hydrodynamic radius, making it ideal for quantifying soluble aggregates and fragments under native conditions. CE-SDS, which separates denatured proteins based on their molecular weight, offers high-resolution purity analysis and detection of fragments and impurities at levels as low as 0.1% [53]. The integration of data from these orthogonal techniques, alongside charge-based separation methods, allows for a thorough understanding of mAb heterogeneity, facilitating robust process development and ensuring product quality.
Methodologies and Principles
Size Exclusion Chromatography (SEC) for Aggregate Analysis
SEC separates molecules based on their hydrodynamic volume as they pass through a porous stationary phase. Larger molecules, such as aggregates, are excluded from the pores and elute first, while smaller fragments and the main monomeric species penetrate the pores to varying degrees and elute later. This size-based separation mechanism makes SEC the gold standard for quantifying aggregates in biopharmaceuticals [54] [55].
Successful SEC method development requires minimizing secondary interactions (electrostatic and hydrophobic) between the protein analyte and the column resin that can compromise the accuracy of quantification. The optimal mobile phase must be systematically determined for each molecule, considering its specific properties.
Key Considerations for SEC Method Development:
- Electrostatic Interactions: Can be mitigated by adjusting the ionic strength of the mobile phase. However, high salt concentrations can induce "salting out" effects, increasing hydrophobic interactions [54].
- Hydrophobic Interactions: Can be minimized by using additives such as organic modifiers (e.g., acetonitrile) or amino acids (e.g., arginine) [54].
- Spatial Aggregation Propensity (SAP): mAbs with a high SAP index are particularly prone to hydrophobic interactions with the stationary phase, often necessitating mobile phase additives like arginine for optimal performance [55].
Capillary Electrophoresis-Sodium Dodecyl Sulfate (CE-SDS) for Purity Assessment
In CE-SDS, proteins are denatured, reduced (or non-reduced), and complexed with SDS, which imparts a uniform negative charge. The complexes are then electrophoretically separated through a polymer network within a capillary. Separation is based on the molecular weight of the polypeptide chains, as the SDS-bound proteins migrate with mobility inversely proportional to the logarithm of their molecular weight [53] [56].
CE-SDS offers significant advantages over traditional SDS-PAGE, including automated operation, quantitative data output, higher resolution, and improved reproducibility. It is widely applied for monitoring purity, integrity, and fragmentation of therapeutic proteins throughout development and quality control [57] [56].
Experimental Protocols
Protocol: SEC Method Development for a mAb with High Aggregation Propensity
This protocol outlines a systematic, Design of Experiments (DoE) approach for developing a robust SEC method for a hydrophobic mAb, based on a published case study [54] [55].
3.1.1 Research Reagent Solutions and Materials
Table 1: Key Reagents and Materials for SEC Method Development
| Item | Function/Purpose |
|---|---|
| TSKgel G3000SWXL Column | Silica-based SEC column for separation by hydrodynamic size. |
| Sodium Phosphate Buffer | Mobile phase component; concentration and pH are critical optimization parameters. |
| L-Arginine Monohydrochloride | Mobile phase additive to minimize hydrophobic interactions with the stationary phase. |
| Sodium Chloride | Used to adjust ionic strength and screen for electrostatic interactions. |
| Acetonitrile | Organic modifier to screen for mitigation of hydrophobic interactions. |
3.1.2 Step-by-Step Procedure
Preliminary Screening:
- Column Selection: Evaluate several commercial SEC columns (e.g., TSKgel G3000SWxl, Bio-Rad BioSil, etc.).
- Mobile Phase Formulation: Test the columns with mobile phases of varying ionic strengths (e.g., 100 mM, 250 mM, and 500 mM sodium phosphate) at a neutral pH.
- Analysis: Inject the mAb of interest and a molecular weight standard. Evaluate chromatograms for peak shape, recovery, and resolution between monomer and aggregate species.
DoE for Optimization:
- Factors: For the selected column, create a DoE matrix to simultaneously optimize two critical parameters: the concentration of sodium phosphate (e.g., 25-100 mM) and the pH (e.g., 6.5-7.8).
- Response Variables: Monitor key responses such as aggregate resolution, peak symmetry, and protein recovery for each run.
Additive Screening:
- If poor peak shape or recovery persists (indicative of secondary interactions), screen mobile phase additives.
- Test arginine (e.g., 100-300 mM), as it is highly effective at disrupting hydrophobic interactions without significantly increasing ionic strength [54] [55].
- As a comparison, test acetonitrile (e.g., 2-10% v/v).
Method Robustness Testing:
- Once optimal conditions are identified (e.g., TSKgel G3000SWXL Column with 25 mM sodium phosphate and 300 mM arginine, pH 7.8), perform a robustness DoE.
- Introduce small, deliberate variations in critical method parameters (e.g., mobile phase pH ±0.1, temperature ±2°C, flow rate ±0.05 mL/min) to establish the method's operational range.
Method Qualification:
- Qualify the final method according to ICH Q2(R1) guidelines, assessing specificity, linearity, accuracy, precision, limit of detection (LOD), and limit of quantitation (LOQ).
The workflow for this systematic development is summarized in the diagram below.
Protocol: CE-SDS Method for Purity Analysis of Protein Antigens
This protocol is adapted from a validated method for the purity analysis of pertussis vaccine antigens (PTx, FHA, PRN) and can be applied to mAbs and other therapeutic proteins [57] [56].
3.2.1 Research Reagent Solutions and Materials
Table 2: Key Reagents and Materials for CE-SDS Analysis
| Item | Function/Purpose |
|---|---|
| CE-SDS Analysis Kit (e.g., AB Sciex) | Provides sample buffer, gel buffer, and MW size standards for reproducible analysis. |
| Bare-Fused Silica Capillary | The separation channel; length and diameter impact resolution. |
| 2-Mercaptoethanol | Reducing agent for reduced CE-SDS analysis. |
| Iodoacetamide | Alkylating agent for non-reduced or carboxymethylation workflows. |
| Centrifugal Filter Device (10 kDa) | For sample desalting and buffer exchange prior to analysis. |
3.2.2 Step-by-Step Procedure
Sample Preparation:
- Desalting: Desalt protein samples (e.g., mAb at 1-2 mg/mL) using a 10 kDa centrifugal filter device.
- Denaturation: Mix the desalted protein with sample buffer containing SDS from the CE-SDS kit.
- Reduction (Optional): For reduced CE-SDS, add 2-mercaptoethanol (e.g., 5% v/v) and heat at 70°C for 5-10 minutes.
- Alkylation (Optional): For non-reduced analysis, iodoacetamide can be added to alkylate free thiols and prevent disulfide scrambling.
Instrumental Setup:
Method Programming:
- Capillary Rinse: Flush the capillary with 0.1 M NaOH, followed by water, and then gel buffer.
- Sample Injection: Perform electrokinetic injection (e.g., 5-10 kV for 1-30 seconds) or pressure injection.
- Separation: Apply a separation voltage of 10-15 kV (reversed polarity, cathode at the detector side). The optimal voltage should be determined to balance resolution and run time.
- Capillary Cleaning: Between runs, rinse the capillary with gel buffer or a prescribed washing solution.
Data Analysis:
- Identify peaks based on migration time compared to an internal SDS-MW standard.
- Calculate the relative percent area of each peak (main peak, fragments, and high molecular weight species) to determine protein purity.
The workflow for CE-SDS analysis is summarized in the diagram below.
Data Presentation and Analysis
SEC Data Interpretation
A well-developed SEC method should provide a chromatogram with baseline resolution between the aggregate, monomer, and fragment peaks. The quantitative data derived from integration is used for stability studies and release testing.
Table 3: Exemplary SEC Data for a mAb Stability Study
| Sample | Aggregate (%) | Monomer (%) | Fragment (%) | Total Recovery (%) |
|---|---|---|---|---|
| Initial (Time 0) | 0.8 | 98.9 | 0.3 | 99.5 |
| 1 Month, 5°C | 0.9 | 98.8 | 0.3 | 99.2 |
| 1 Month, 25°C | 1.5 | 97.8 | 0.7 | 98.1 |
| 3 Months, 25°C | 2.3 | 96.5 | 1.2 | 97.2 |
| Acceptance Criteria | ≤2.0% | ≥97.0% | ≤1.5% | ≥95.0% |
CE-SDS Data Interpretation
CE-SDS provides a high-resolution profile of the protein's purity, revealing the main band and low-abundance impurities like fragments and clipped species. The method's precision for purity and molecular weight determination is typically below 10% RSD [57].
Table 4: CE-SDS Purity and Molecular Weight Data for a Model mAb (n=3)
| Species | Theoretical MW (kDa) | Observed MW (kDa) ± RSD | Purity (%) ± RSD |
|---|---|---|---|
| Heavy Chain | 50.0 | 50.2 ± 0.4% | - |
| Light Chain | 25.0 | 25.1 ± 0.6% | - |
| Non-Glycosylated H.C. | 48.2 | 48.3 ± 0.5% | 1.2 ± 5.1% |
| Main Species (Intact mAb) | 148.1 | 149.5 ± 0.3% | 96.5 ± 0.8% |
| Fragment 1 | ~45.0 | 45.5 ± 1.1% | 1.5 ± 6.3% |
| LOQ | - | - | 0.2% |
Integration with Charge Variant Analysis
The characterization of mAbs is incomplete without assessing charge variants, which are critical CQAs. Capillary Zone Electrophoresis (CZE) is a high-efficiency tool for separating charge variants such as deamidated species, C-terminal lysine variants, and sialic acid variants [5]. The data from SEC (aggregates), CE-SDS (purity/fragments), and CZE (charge variants) together form a comprehensive orthogonal analytical package.
For instance, a deamidation event, which introduces a negative charge detectable by CZE, might also promote aggregation under certain conditions, which would be monitored by SEC. Similarly, fragments detected by CE-SDS may have different charge profiles than the intact molecule. Integrating these data sets allows scientists to build a holistic understanding of the product's stability, manufacturability, and ultimately, its safety and efficacy profile.
Monoclonal antibodies (mAbs) are complex biopharmaceutical products exhibiting significant microheterogeneity due to post-translational modifications (PTMs) that occur during manufacturing and storage [12]. Charge variant analysis is a critical component of biotherapeutic characterization mandated by regulatory guidelines to ensure product safety, efficacy, and quality [36]. These charge variants can impact critical quality attributes (CQAs) including stability, potency, and serum half-life [3].
This case study demonstrates how an integrated multi-method workflow addresses characterization gaps for a therapeutic mAb by combining orthogonal separation techniques with advanced detection technologies. We present a comprehensive approach to identify, quantify, and characterize charge variants that closes technical gaps left by single-method approaches, enabling more effective control strategy implementation for commercial mAb products [58].
Experimental Design and Workflow Integration
Workflow Strategy
The characterization workflow employs orthogonal techniques at multiple structural levels (intact, subunit, and peptide) to comprehensively map charge heterogeneity. This approach leverages the complementary strengths of each method while mitigating their individual limitations [5] [12] [58].
Research Reagent Solutions
Table 1: Essential research reagents and materials for charge variant characterization
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Capillary Coatings | Controls electroosmotic flow, prevents protein adsorption | Successive multiple ionic-polymer layers (SMIL), diethylaminoethyl-dextran–poly(sodium styrene sulfonate) [5] |
| MS-Compatible BGE | Enables direct CZE-MS coupling while maintaining separation | Acidic background electrolytes with volatile additives [5] |
| Reference Standards | System suitability monitoring and method qualification | USP mAb Reference Standards (e.g., USP mAb003) [5] [59] |
| Enzymes | Targeted proteolysis for subunit and peptide analysis | Carboxypeptidase B (C-terminal Lys removal), sialidase (sialic acid removal), trypsin [3] [12] |
| pH Gradient Buffers | Charge-based separation in ion-exchange chromatography | Commercial pH gradient buffer kits for linear pH gradients [36] |
Materials and Methods
Capillary Zone Electrophoresis with UV Detection (CZE-UV)
Principle: Separates intact mAb charge variants based on differences in their charge-to-size ratios under acidic conditions [12].
- Capillary: Fused silica, 50/100 µm inner diameter, 40-60 cm length
- Coating: Dynamic neutral coating or SMIL coating with diethylaminoethyl-dextran–poly(sodium styrene sulfonate) [5]
- Background Electrolyte: Acidic volatile electrolytes (e.g., formic acid, acetic acid)
- Sample Preparation: mAb solution at 1 mg/mL in BGE
- Injection: 40 mbar for 5 seconds
- Separation Voltage: ±10-30 kV (polarity depends on coating)
- Detection: UV at 214 nm
This method provides high-resolution separation of mAb charge variants including C-terminal lysine variants, deamidated species, and other proteoforms [5].
CZE Coupled to Mass Spectrometry (CZE-ESI-MS)
Principle: Combines high-efficiency charge-based separation with mass determination for direct variant identification [5].
- Interface: Sheathless or sheath-liquid ESI interface
- Capillary Coating: Cationic successive multiple ionic-polymer layers (SMIL)
- BGE: 50 mM acetic acid (pH 3.0) or other volatile acids
- MS Parameters: Positive ion mode, mass range 500-4000 m/z
- Application: Direct characterization of proteoforms including lysine variants, glycosylated forms, and acidic variants
This approach enables separation of variants with small mass differences (e.g., deamidation +1 Da, disulfide bridge reduction +2 Da) that cannot be resolved by MS alone [5].
Ion-Exchange Chromatography (IEC) with Fraction Collection
Principle: Separates charge variants based on electrostatic interactions with stationary phase using pH or salt gradients [36] [31].
- Column: Strong cation exchange (e.g., ProPac SCX-10)
- Mobile Phase A: Low pH buffer (e.g., 20 mM sodium phosphate, pH 6.0)
- Mobile Phase B: High pH buffer or salt gradient
- Gradient: Linear pH gradient from pH 6.0 to 9.5 over 30 minutes
- Flow Rate: 0.5-1.0 mL/min
- Detection: UV at 280 nm
- Fraction Collection: Semi-preparative scale for variant isolation
pH gradients offer advantages over salt gradients including global applicability to different mAbs and better MS compatibility [36].
Multi-Attribute Method (MAM) with LC-MS
Principle: Uses peptide mapping with mass spectrometry to directly monitor multiple PTM CQAs simultaneously [58].
- Digestion: Tryptic digestion under denaturing and reducing conditions
- LC Separation: Reversed-phase chromatography with C18 column
- MS Analysis: High-resolution mass spectrometry (e.g., QDa mass spectrometer)
- Data Analysis: Targeted monitoring of specific PTMs (deamidation, oxidation, glycation) and new peak detection
This method enables replacement of traditional profile-based methods with direct CQA monitoring [58].
Results and Data Interpretation
Charge Variant Separation and Identification
The multi-method workflow successfully resolved and identified critical charge variants in the therapeutic mAb. Each technique provided complementary information for comprehensive characterization.
Table 2: Charge variant identification across analytical platforms
| Variant Type | CZE-UV Migration | CZE-MS Mass Shift | IEC Elution | Potential Modifications |
|---|---|---|---|---|
| Acidic 1 | 19.8 min (basic) | +1 Da | Early eluting | Deamidation (CDR), glycation [58] |
| Acidic 2 | 21.1 min (main) | +1 Da | Middle acidic | Deamidation, sialic acid [31] |
| Acidic 3 | 22.5 min (acidic) | 0 Da | Late acidic | Fragmentation, trisulfide bonds [31] |
| Main | 21.1 min | 0 Da | Center peak | Glycosylated, unmodified [58] |
| Basic 1 | 19.8 min | 0 Da | Early basic | C-terminal Lys, N-terminal Glu [31] |
| Basic 2 | 18.5 min | +16 Da | Late basic | Methionine oxidation, aggregates [58] [31] |
Quantitative Charge Variant Distribution
The relative abundance of each charge variant was quantified across multiple analytical techniques, demonstrating consistent pattern recognition despite different separation mechanisms.
Table 3: Quantitative distribution of charge variants across platforms
| Variant | CZE-UV Area % | IEC Area % | CZE-MS Relative Abundance | Biological Potency |
|---|---|---|---|---|
| Acidic 1 | 1.1% | 1.1% | 0.9% | <25% [58] |
| Acidic 2 | 6.7% | 6.7% | 7.1% | 33% [58] |
| Acidic 3 | 9.7% | 9.7% | 10.2% | 36% [58] |
| Main | 79.7% | 79.7% | 78.5% | 98% [58] |
| Basic | 2.8% | 2.8% | 3.3% | 78% [58] |
Structural Characterization of Variants
Advanced mass spectrometry techniques identified specific PTMs responsible for charge differences and their potential impact on biological activity.
- C-terminal Lysine Variants: Identified as basic variants showing mass differences corresponding to presence of 0, 1, or 2 lysine residues [5] [31]
- Deamidation Products: Detected as acidic variants with +1 Da mass increase, particularly in complementarity-determining regions (CDR) with significant potency impact [58]
- Oxidation Variants: Basic variants showing +16 Da mass increase from methionine oxidation, affecting FcRn binding [31]
- Glycation Products: Acidic variants with mass increases corresponding to glucose adducts [58]
Advanced Protocol: CZE-ESI-MS for Intact mAb Characterization
Capillary Coating Protocol (SMIL Coating)
The successive multiple ionic-polymer layer (SMIL) coating is critical for achieving high-resolution separations [5].
Capillary Pretreatment:
- Flush new capillary with 1 M NaOH for 10 minutes
- Rinse with ultrapure water (UPW) for 5 minutes
- Equilibrate with 20 mM HEPES solution (pH 7.4) for 10 minutes
Layer-by-Layer Assembly:
- Flush with polycation solution (3 g/L in HEPES) for 7 minutes
- Rinse with HEPES solution for 3 minutes
- Flush with polyanion solution (3 g/L in HEPES) for 7 minutes
- Rinse with HEPES solution for 3 minutes
- Repeat until 5 total layers are deposited
- Wait 5 minutes for layer stabilization
Final Equilibration:
- Rinse with UPW for 3 minutes
- Equilibrate with BGE for 10 minutes
- Store capillary in BGE between runs
CZE-ESI-MS Analysis Conditions
Separation Conditions:
- BGE: 50 mM acetic acid (pH 3.0)
- Capillary: SMIL-coated, 60 cm length × 50 μm ID
- Voltage: -10 kV (cathodic at inlet)
- Temperature: 25°C
- Injection: 40 mbar × 5 seconds (1 mg/mL mAb in BGE)
MS Detection Conditions:
- Ionization: ESI positive mode
- Drying Gas: 4 L/min, 200°C
- Nebulizer: 5 psi
- Capillary Voltage: 4000 V
- Mass Range: 500-4000 m/z
This protocol enables separation of C-terminal lysine variants from the main form, as well as several acidic variants and monoglycosylated mAb forms [5].
Discussion
Method Complementarity and Gap Closure
The multi-method workflow effectively closes characterization gaps through orthogonal technique integration:
- CZE-UV provides high-resolution separation with robustness and reproducibility for routine analysis [12]
- CZE-ESI-MS enables direct variant identification with molecular weight information [5]
- IEC with fractionation allows isolation of variants for further functional characterization [58]
- MAM with LC-MS offers specific monitoring of CQAs for control strategy implementation [58]
The combination of these methods addresses limitations of individual techniques, particularly regarding variant identification, quantification, and criticality assessment.
Impact on Control Strategy
Implementation of this workflow enabled replacement of traditional profile-based IEC methods with a targeted CQA monitoring approach for the commercial mAb product [58]. The enhanced characterization demonstrated that:
- Direct monitoring of specific CQAs (CDR deamidation, oxidation, glycation) via MAM provides more meaningful quality control than profile-based methods
- SE-HPLC and CE-SDS effectively control for non-PTM charge variants (HMWS, LMWS)
- The combination of MAM, SE-HPLC, and CE-SDS ensures comprehensive control of all critical charge variants
This strategy has received regulatory approval from multiple health authorities for commercial testing [58].
This case study demonstrates that a carefully designed multi-method workflow effectively closes characterization gaps for mAb charge variants. The integration of CZE-UV, CZE-ESI-MS, IEC, and MAM provides comprehensive structural information that enables science-based control strategy implementation. The orthogonal nature of these techniques ensures complete variant characterization while mitigating the limitations of individual methods.
This approach supports regulatory submissions by providing thorough product understanding and facilitates implementation of targeted control strategies focused on direct monitoring of critical quality attributes. The workflow can be adapted for various biotherapeutic proteins including antibody-drug conjugates, bispecific antibodies, and biosimilar products during development and commercialization.
From Analysis to Control: Strategies for Optimizing and Troubleshooting Charge Profiles
The comprehensive characterization of charge variants is a critical requirement in the development of therapeutic monoclonal antibodies (mAbs). While regulatory mandates necessitate thorough analysis, a significant characterization gap persists for acidic species, with studies reporting that approximately one-third of these variants remain unidentified in standard analyses. This application note details integrated strategies and protocols to address these challenges, leveraging advanced fractionation techniques and multi-analytical approaches to resolve unidentified acidic species, ensure product quality, and meet regulatory expectations for biopharmaceutical development.
Therapeutic monoclonal antibodies exhibit inherent heterogeneity due to enzymatic and chemical post-translational modifications (PTMs) occurring during manufacturing and storage [37] [26]. These modifications generate charge variants, which are typically categorized as acidic, main, or basic species based on their isoelectric points (pI) or chromatographic elution profiles relative to the main antibody species [37]. Monitoring these variants is crucial as they can impact critical quality attributes (CQAs), including stability, biological activity, and immunogenicity [60] [61].
Despite advances in analytical technologies, a substantial characterization gap persists. A case study on a recombinant IgG1 (mAb1) revealed that initial characterization could only account for approximately 65% of its acidic species, leaving about 35% unidentified [26]. This gap is not uncommon and underscores the analytical challenges in achieving a complete characterization, potentially leaving impactful modifications undetected. This document outlines practical strategies and detailed protocols to bridge this gap, enabling a more comprehensive analysis of acidic charge variants.
Quantitative Analysis of Acidic Species Modifications
A thorough characterization begins with understanding the known contributors to acidic species. The following table summarizes common PTMs leading to acidic variant formation and their prevalence as identified in a detailed characterization study [26].
Table 1: Common Modifications in Acidic Species and Their Quantitative Distribution in a Representative mAb1 Study
| Modification Type | Reported Prevalence in mAb1 Acidic Species (%) | Primary Analytical Method(s) for Detection |
|---|---|---|
| Deamidation | 17.2 | Peptide Mapping, LC-MS/MS |
| Glycation | 15.2 | Intact Mass Analysis, Boronate Affinity Chromatography |
| Oxidation | 21.6 | Peptide Mapping, Intact Mass Analysis |
| Low Molecular Weight Species (Fragments) | 10.0 | SEC, CE-SDS |
| Fab Glycan | 0.6 | Peptide Mapping, LC-MS/MS |
| Hydroxylation | 0.6 | Peptide Mapping |
| Unidentified Species | ~34.0 | Multi-pronged Investigation Required |
The complexity of acidic species arises from the diversity of modifications. Key contributors include:
- Deamidation: The conversion of asparagine (Asn) to aspartic acid (Asp) or isoaspartic acid (isoAsp), introducing a negative charge. This occurs frequently in flexible regions like the complementarity-determining regions (CDRs) and constant domains [37] [61].
- Glycation: The non-enzymatic addition of glucose to lysine residues, which can be quantified at the intact level or via peptide mapping [37] [26].
- Sialylation: The attachment of sialic acid residues to glycan structures, adding significant negative charge [37].
- Other Modifications: Trisulfide bonds, cysteinylation, thiosulfide modifications, and non-classical disulfide linkages have also been reported [37].
Experimental Protocols for Comprehensive Characterization
Strategic Workflow for Acidic Species Identification
A systematic approach is essential for isolating and characterizing acidic variants. The following workflow diagram outlines the key stages, from initial separation to in-depth analysis of unidentified species.
Protocol 1: Fractionation of Charge Variants by Free-Flow Electrophoresis (FFE)
Principle: FFE continuously separates protein samples in a fluid curtain based on their isoelectric points under an electric field, enabling preparative-scale fractionation of charge variants under native conditions [60].
Materials:
- Instrument: Free-flow electrophoresis system (e.g., from FFE service, Gmbh).
- Separation Buffer: Contains carrier ampholytes to establish a stable pH gradient.
- Sample: Monoclonal antibody at 10-50 mg/mL in a low-salt buffer.
- Collection Plates: 96-well plates for automated fraction collection.
- Purification: Protein A cartridges for post-FFE purification of fractions.
Procedure:
- Sample Preparation: Dilute the mAb sample with separation buffer to a final conductivity of < 2 mS/cm. Centrifuge at 10,000 × g for 5 minutes to remove particulates.
- FFE System Setup: Prime the separation chamber with separation buffer. Set the following parameters:
- Hydrodynamic Flow Rate: 1.5 - 3.0 mL/h per fraction
- Electric Field Strength: 500 - 1000 V
- Cooling Temperature: 4-10 °C
- pH Gradient: e.g., pH 5-9, established by ampholytes
- Separation Run: Continuously inject the prepared sample into the separation chamber. The applied electric field will focus charge variants into distinct streams based on pI.
- Fraction Collection: Collect the separated streams into a 96-well plate. Typically, 96 fractions are collected across the entire pH range.
- Fraction Purification and Analysis: Pass fractions through protein A cartridges to remove ampholytes and buffer components. Concentrate if necessary. Assess the purity and identity of each fraction by imaged capillary isoelectric focusing (icIEF) [60].
Protocol 2: In-Depth Characterization of FFE Fractions
Principle: Isolated and purified fractions are subjected to a suite of analytical techniques to identify modifications and structural changes.
Materials:
- LC-MS System: Ultra-high-performance liquid chromatography coupled to a high-resolution mass spectrometer.
- Enzymes: Lysyl endopeptidase (Lys-C) and/or trypsin for peptide mapping.
- SEC Columns: For aggregate analysis.
- HDX-MS System: For higher-order structure analysis.
Procedure:
- Intact Mass Analysis:
- Objective: Determine molecular weight shifts to identify modifications like glycation (+162 Da) or sialylation.
- Method: Inject ~2 µg of purified FFE fraction onto a reversed-phase UHPLC column coupled to the MS. Use a shallow acetonitrile gradient with 0.1% formic acid for desalting and separation. Perform data deconvolution to determine the intact mass [60] [26].
Peptide Mapping with LC-MS/MS:
- Objective: Locate site-specific modifications (deamidation, oxidation, etc.).
- Method: Denature and reduce the antibody from FFE fractions. Digest with Lys-C (or trypsin) at 37°C for 4-6 hours. Analyze the resulting peptides using LC-MS/MS with a C18 column. Use data-dependent acquisition to fragment peptides for PTM identification [61] [26].
Higher-Order Structure (HOS) Analysis:
- Objective: Detect conformational differences that may not be explained by known PTMs.
- Method (HDX-MS): Dilute the FFE fraction in D₂O buffer and incubate for various time points (e.g., 10s, 1min, 10min, 1h). Quench the reaction and digest the protein on ice. Analyze the peptides by LC-MS to measure deuterium uptake rates. Significant differences in uptake between acidic and main species indicate conformational changes [26].
Aggregation and Fragmentation Analysis:
- Objective: Quantify fragments and aggregates that may co-elute with acidic species.
- Method: Analyze FFE fractions by Size-Exclusion Chromatography (SEC) using a compatible mobile phase (e.g., PBS, pH 6.8). Monitor UV absorbance at 280 nm. Peaks eluting before the main peak are aggregates; peaks eluting after are fragments [60] [19].
The Scientist's Toolkit: Research Reagent Solutions
Successful characterization relies on a suite of specialized reagents and instruments. The following table details key solutions for addressing acidic variant characterization gaps.
Table 2: Essential Research Reagents and Tools for Acidic Species Characterization
| Tool / Reagent | Primary Function | Application Context in Characterization |
|---|---|---|
| Linear pH Gradient Buffers | Enables robust, generic cation-exchange chromatography (CEX) for mAb charge variant separation. | Used for initial profiling and preparative fractionation of acidic, main, and basic species [36]. |
| Strong Cation-Exchange (SCX) Columns | High-resolution separation of charge variants based on surface charge distribution. | Critical for isolating pure acidic species fractions for subsequent analysis (e.g., ProPac SCX-10) [36] [61]. |
| Free-Flow Electrophoresis (FFE) | Continuous, preparative-scale fractionation of proteins based on pI under native conditions. | Overcomes limitations of column-based methods; allows collection of sufficient material for multiple orthogonal assays [60]. |
| High-Resolution Mass Spectrometer | Provides accurate molecular weight determination and identifies modifications via intact mass and peptide mapping. | Core tool for identifying mass shifts (+162 for glycation, +1 for deamidation) and locating modification sites [60] [26]. |
| Hydrogen-Deuterium Exchange (HDX) MS | Probes protein higher-order structure and conformational dynamics in solution. | Investigates potential conformational differences in unidentified acidic species that lack a clear chemical modification signature [26]. |
| Capillary Isoelectric Focusing (icIEF) | High-resolution analytical technique to measure charge heterogeneity based on pI. | Used for purity assessment of collected fractions and for routine, high-throughput charge variant analysis [36] [61]. |
The challenge of unidentified acidic species in monoclonal antibodies demands a systematic and multi-faceted analytical strategy. By integrating advanced separation technologies like FFE with deep characterization tools such as HDX-MS and rigorous intact mass analysis, researchers can close the quantification gaps that persist in standard workflows. The protocols and strategies detailed in this application note provide a actionable framework for achieving a more complete understanding of charge heterogeneity, ultimately ensuring the development of safe, efficacious, and high-quality biotherapeutic products.
Forced Degradation Studies to Enrich and Identify Low-Abundance Variants
Forced degradation studies are an integral component in the development of therapeutic monoclonal antibodies (mAbs), serving to intentionally expose these complex molecules to exaggerated stress conditions. The primary objective is to accelerate degradation pathways, thereby enriching low-abundance product variants that are typically present at minimal levels in drug substances and products [62]. These studies provide a critical proactive assessment of a molecule's intrinsic stability, revealing potential degradation products that could impact drug safety and efficacy [62] [63].
Within the broader context of charge variant analysis, forced degradation provides a controlled mechanism to generate and study acidic and basic species, enabling researchers to establish comprehensive impurity profiles and validate stability-indicating methods essential for regulatory compliance [37] [64]. By understanding the specific modifications that generate charge heterogeneity—such as deamidation, oxidation, and glycation—scientists can make informed decisions throughout the product lifecycle, from candidate selection to commercial manufacturing [62] [37].
Purposes and Applications in mAb Development
Forced degradation studies serve multiple strategic purposes throughout the biopharmaceutical development lifecycle, extending far beyond simple stability assessment.
Table 1: Applications of Forced Degradation Studies in mAb Development
| Application | Purpose and Rationale |
|---|---|
| Manufacturability Assessment | Evaluates the intrinsic stability of multiple candidates under relevant process conditions to select the most stable molecule for development [62]. |
| Formulation Development | Identifies optimal buffer compositions, excipients, and pH conditions that provide appropriate long-term stability [62]. |
| Stability-Indicating Method Development | Using degraded samples to establish analytical methods that can monitor degradation throughout the product shelf life [62] [63]. |
| Critical Quality Attributes (CQA) Assessment | Generates specific modifications at higher abundance to facilitate identification and monitoring of CQAs [62]. |
| Comparability Assessment | Reveals differences in degradation profiles and kinetics between pre- and post-change materials that may not be detectable by routine testing [62] [65]. |
| Impurity Isolation and Characterization | Provides sufficient quantities of low-abundance variants for detailed structural and functional analysis [62] [64]. |
The data generated from these studies forms the scientific foundation for regulatory submissions, demonstrating a comprehensive understanding of product stability and degradation pathways as expected by agencies such as the FDA and EMA [62] [63]. Furthermore, forced degradation helps define the boundary of instability under various environmental factors, supporting risk assessment for accidental excursions during shipping and handling [62].
Common Degradation Pathways and Conditions
The selection of appropriate stress conditions is guided by the likelihood of mAbs being exposed to these factors during manufacturing, storage, and administration. Each stress condition preferentially accelerates specific degradation pathways.
Table 2: Common Forced Degradation Conditions and Their Primary Effects on mAbs
| Stress Condition | Major Degradation Pathways | Key Influencing Factors |
|---|---|---|
| High Temperature (e.g., 40-50°C) | Aggregation (soluble/insoluble), fragmentation (hinge region), deamidation, oxidation, aspartate isomerization, disulfide scrambling [62] [65] [66] | pH, buffer composition, protein concentration, temperature [62] |
| Freeze-Thaw Cycling | Formation of non-covalent aggregates (dimers, multimers), precipitation [62] | Cooling/warming rates, pH, excipients, protein concentration [62] |
| Agitation (shaking/stirring) | Insoluble and soluble aggregation (covalent/non-covalent), non-native disulfide bonds [62] | Headspace, interface interactions, presence of surfactants, container type [62] |
| Low/High pH (e.g., pH 3-10) | Fragmentation, deamidation, disulfide bond scrambling, thioether formation [62] [37] | Buffer species, incubation time, temperature [62] |
| Oxidation (e.g., H₂O₂) | Methionine and tryptophan oxidation, potentially affecting biological activity [62] [64] | Oxidant concentration, exposure duration, presence of catalysts [62] |
| Light Exposure | Photo-degradation, potentially leading to aggregation and fragmentation [62] [63] | Light intensity, wavelength spectrum, container transparency [62] |
The nonglycosylated mAb variant deserves special consideration in thermal stress studies, as it demonstrates compromised thermal stability and increased propensity to form large aggregates at elevated temperatures commonly used in forced degradation studies (40-50°C) [66]. This phenomenon highlights the importance of understanding how specific product-related variants can disproportionately influence the overall degradation profile of mAb products under stress conditions.
Diagram 1: Forced Degradation Pathways and Analytical Detection in mAbs. This workflow illustrates how different stress conditions induce specific degradation pathways, resulting in variant species detectable by orthogonal analytical methods. LC-MS/MS provides detailed characterization for multiple variant types.
Experimental Protocols for Forced Degradation
Thermal Stress Protocol
Objective: To accelerate degradation pathways relevant to long-term storage, including aggregation, fragmentation, and deamidation [62] [65].
Materials:
- mAb drug substance or drug product
- Controlled temperature incubators (40°C, 50°C)
- Appropriate container closure system
Procedure:
- Prepare mAb samples at target protein concentration in formulation buffer.
- Aliquot samples into suitable containers (e.g., glass vials).
- Place samples in incubators maintained at 40°C and 50°C [65] [66].
- Include control samples stored at recommended storage temperature (e.g., 5°C).
- Remove samples at predetermined time points (e.g., 3, 7, and 14 days) [65].
- Analyze samples alongside controls using appropriate analytical methods.
Key Considerations:
- Temperature selection should be below the melting temperature (Tm) of the mAb to avoid non-representative unfolding [66].
- The nonglycosylated variant may preferentially aggregate at temperatures of 45°C and above [66].
- Include multiple time points to establish degradation kinetics.
pH-Based Stress Protocol
Objective: To generate charge variants through deamidation and fragmentation, particularly enriching acidic species [62] [37].
Materials:
- mAb drug substance
- Buffers covering pH range 3-10 (e.g., citrate, phosphate, Tris)
- Purification equipment for buffer exchange (if needed)
Procedure:
- Prepare mAb samples in buffers at different pH values (e.g., pH 4, 7, 9).
- Adjust protein concentration to 1-10 mg/mL.
- Incubate samples at 25-40°C for 1-14 days [62].
- Terminate reactions by adjusting to formulation pH or through buffer exchange.
- Analyze samples for charge variants and fragments.
Key Considerations:
- Fragmentation is highly pH-dependent, with minimal levels around pH 6 and acceleration at both low and high pH [62].
- High pH conditions (e.g., pH 9) can trigger disulfide bond scrambling and thioether formation [62].
- Control samples should be maintained in formulation buffer at recommended pH.
Oxidative Stress Protocol
Objective: To generate oxidized species, particularly methionine and tryptophan residues, for identification and monitoring [62] [64].
Materials:
- mAb drug substance
- Hydrogen peroxide (H₂O₂) solution
- Buffer for dilution
Procedure:
- Prepare mAb sample in appropriate buffer.
- Add H₂O₂ to final concentrations ranging from 0.01-0.1%.
- Incubate at 25-37°C for 1-24 hours [62].
- Quench reaction by desalting or addition of catalase.
- Analyze for oxidation products and changes in biological activity.
Key Considerations:
- Tert-butyl hydroperoxide can be used as an alternative oxidant.
- Site-specific oxidation may occur depending on solvent accessibility.
- Oxidation in the Fc region may affect FcRn binding, potentially influencing serum half-life [64].
Analytical Characterization of Stress-Generated Variants
Charge Variant Analysis by Cation Exchange Chromatography (CEX)
Principle: Separates mAb variants based on differences in surface charge, effectively resolving acidic species, main species, and basic species [36] [37].
Protocol:
- Column: Use strong cation exchange (SCX) column such as ProPac SCX-10 [36].
- Mobile Phase:
- Buffer A: 10-20 mM sodium phosphate, pH 6.0-7.0
- Buffer B: A + 0.2-0.5 M NaCl or pH gradient elution
- Gradient: Linear salt or pH gradient over 30-90 minutes [36] [37].
- Detection: UV at 280 nm.
- Fraction Collection: Collect peaks for further characterization.
Applications:
- Resolves variants with modifications that alter charge (deamidation, sialylation, C-terminal lysine) [37].
- Acidic species typically elute earlier than main peak in CEX [37].
- Basic species typically elute later than main peak in CEX [37].
Size Variant Analysis by Size Exclusion Chromatography (SEC)
Principle: Separates mAb variants based on hydrodynamic size, resolving high molecular weight (HMW) aggregates, monomer, and low molecular weight (LMW) fragments [64] [66].
Protocol:
- Column: Use SEC column with appropriate separation range (e.g., Acquity BEH200 SEC) [66].
- Mobile Phase: 150 mM ammonium acetate or phosphate buffer with 150-200 mM NaCl, pH 6.0-7.0.
- Flow Rate: 0.2-1.0 mL/min depending on column dimensions.
- Detection: UV at 280 nm.
- Advanced Coupling: For detailed characterization, couple with multi-angle light scattering (MALS) or mass spectrometry (MS) [66].
Applications:
- Quantification of soluble aggregates and fragments [64].
- SEC-UV is stability-indicating but does not provide detailed characterization of aggregates without orthogonal techniques [66].
Peptide Mapping with LC-MS/MS for Structural Characterization
Principle: Provides site-specific identification of post-translational modifications and degradation products through proteolytic digestion and mass spectrometric analysis [65] [64].
Protocol:
- Denaturation: Incubate mAb in 6 M guanidine HCl or 8 M urea.
- Reduction: Add dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP).
- Alkylation: Add iodoacetamide or iodoacetic acid.
- Digestion: Use trypsin or other proteases at enzyme-to-substrate ratio of 1:20-1:50.
- LC-MS/MS Analysis:
- Column: Reversed-phase C18 column (1.0 × 150 mm, 1.7-3.0 μm)
- Mobile Phase: A: 0.1% formic acid in water; B: 0.1% formic acid in acetonitrile
- Gradient: 5-40% B over 60-120 minutes
- Mass Spectrometer: High-resolution tandem mass spectrometer
Applications:
- Identification of specific deamidation sites (e.g., in CDR regions) [37].
- Localization of oxidation to specific methionine or tryptophan residues [64].
- Detection of glycation, isomerization, and other chemical modifications [37] [64].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Forced Degradation Studies
| Category | Specific Items | Function and Application |
|---|---|---|
| Chromatography Columns | ProPac SCX-10 (cation exchange), Acquity BEH200 SEC (size exclusion), reversed-phase C4/C8/C18 [36] [66] | Separation of charge variants, size variants, and hydrophobic species |
| Enzymes and Reagents | Trypsin/Lys-C, carboxypeptidase B, sialidase, PNGase F, dithiothreitol (DTT), iodoacetamide [37] [64] | Protein digestion for peptide mapping, removal of specific modifications (C-terminal lysine, sialic acid) |
| Stress Agents | Hydrogen peroxide, hydrochloric acid, sodium hydroxide, polysorbates [62] [67] | Induction of oxidative, acidic, and basic stress; prevention of interfacial stress |
| Buffers and Chemicals | Ammonium acetate, sodium phosphate, formic acid, acetonitrile (LC-MS grade) [65] [66] | Mobile phase preparation, sample preparation, MS-compatible solvents |
| Analytical Standards | NISTmAb reference material, in-house reference standards [36] | System suitability testing, method qualification, data benchmarking |
Forced degradation studies represent a powerful approach for enriching and identifying low-abundance variants in therapeutic monoclonal antibodies. By employing a systematic combination of stress conditions and orthogonal analytical methods, scientists can comprehensively characterize charge variants and other product-related species that may impact drug quality, safety, and efficacy. The protocols outlined in this application note provide a framework for designing studies that meet regulatory expectations while advancing the understanding of mAb stability. As the biopharmaceutical landscape continues to evolve with increasingly complex modalities, forced degradation studies will remain essential for ensuring the development of safe, effective, and high-quality biologic therapeutics.
Charge heterogeneity in monoclonal antibodies (mAbs), primarily caused by post-translational modifications (PTMs), represents a significant challenge in ensuring the consistency, stability, and efficacy of therapeutic products [18]. These charge variants, classified as acidic, main, or basic species, are considered Critical Quality Attributes (CQAs) as they can impact the structural integrity, biological activity, pharmacokinetics, and overall safety profiles of biotherapeutics [18]. The upstream process, particularly the selection of appropriate cell lines and the formulation of culture media and additives, plays a fundamental role in modulating these charge profiles [68]. This document provides detailed application notes and experimental protocols for optimizing these upstream elements to control charge variants, framed within the broader context of charge variant analysis research.
The Impact of Media and Additives on Charge Variants
Cell culture media composition directly influences cellular metabolism and the subsequent PTMs that drive charge heterogeneity [18]. Specific medium components and process parameters can either amplify or diminish undesirable modifications.
- Acidic Variants are often generated through deamidation of asparagine residues, sialylation of glycans, or glycation processes, which increase negative charge or mask positive charges [68] [18]. These are often accelerated by high pH, elevated temperature, and long culture durations [18].
- Basic Variants typically arise from incomplete removal of C-terminal lysine, incomplete formation of N-terminal pyroglutamate, or succinimide formation, leading to a more positive charge [18].
Systematic optimization of media and feeds is therefore critical for controlling the distribution of these variants. The S-OptiCharge platform exemplifies a structured approach, employing methodical screening of additives and media combined with a Design of Experiments (DoE) methodology to optimize the main peak in a molecule's charge profile, as confirmed by cation-exchange chromatography (CEX) [68].
Table 1: Key Post-Translational Modifications Affecting mAb Charge and Their Drivers
| Charge Variant | Common PTMs | Key Influencing Factors |
|---|---|---|
| Acidic | Deamidation (Asn → Asp/isoAsp), Sialylation, Glycation, Oxidation (Trp) [18] | High pH, high temperature, long culture duration, oxidative stress, nutrient depletion [18] |
| Main | N-terminal pyroglutamate formation, complete C-terminal lysine removal, core neutral N-glycosylation [18] | Optimal biosynthesis, controlled process conditions, efficient enzymatic processing [18] |
| Basic | Incomplete C-terminal lysine removal, incomplete N-terminal pyroGlu formation, succinimide formation, C-terminal amidation [18] | Suboptimal enzymatic processing, low pH, cell line-specific enzyme levels [18] |
Experimental Protocols
Protocol 1: Systematic Screening of Media Additives for Charge Profile Optimization
This protocol outlines a stepwise procedure for identifying media additives that improve the main peak proportion and reduce acidic variants in mAbs produced by CHO cells [68].
Materials and Equipment
- Cell Line: A CHO cell line expressing the mAb of interest [68].
- Basal and Feed Media: Commercially available or proprietary formulations [68].
- Test Additives: A library of candidate chemicals, vitamins, and nutrients (e.g., 10 initial candidates) [68].
- Equipment: Bioreactor or shake flask systems, CEX-HPLC system for charge variant analysis, cell culture incubator [68].
Procedure
- Preparation: Adapt the production CHO cell line to the basal media to be used in the screening.
- Initial Additive Screening:
- Supplement the basal media with each of the candidate additives.
- Inoculate cells at a density of ~0.5 × 10^6 cells/mL in 14-day batch cultures. Add feed media on days 3, 5, and 7 [68].
- Harvest the culture and purify the mAb.
- Charge Variant Analysis:
- Analyze the purified mAb samples using CEX-HPLC to determine the distribution of acidic, main, and basic variants [68].
- Identify additives that yield a statistically significant increase in the main peak and a reduction in the acidic peak compared to a negative control (basal media without additive) [68].
- Secondary Screening:
- Narrow down the top-performing additives (e.g., 4 from the initial 10) and test them in combination with different basal and feed media [68].
- Compare the results to select the single most effective additive and media combination for the specific molecule.
Data Analysis
Compare the CEX chromatograms to quantify the percentage of acidic, main, and basic species. Statistical analysis (e.g., t-test) should confirm the significance of improvements.
Protocol 2: Cell Line Screening for Optimal Charge Profiles
This protocol describes the screening of different host cell lines to identify a clone that naturally produces a more favorable charge variant profile [68].
Materials and Equipment
- Host Cell Lines: Multiple CHO cell variants (e.g., CHO-K1, CHO-S, CHO-DG44) [68] [69].
- Media: A standardized, optimized basal and feed media combination.
- Equipment: Ambr250 or similar micro-bioreactor system for high-throughput screening, CEX-HPLC, cell counter and viability analyzer [69].
Procedure
- Transfection and Pool Generation: Stably transfect the mAb gene of interest into the different CHO host cell lines using a system like glutamine synthetase (GS) or DHFR [69].
- Initial Clone Screening:
- Screen a large number of clones from each host cell line in a 96-well micro-bioreactor format.
- Evaluate cell growth, viability, and titer to down-select to a manageable number of high-producing clones from each host [69].
- Charge Profile Analysis:
- Produce mAbs from the down-selected clones in a fed-batch process.
- Purify the mAbs and analyze the charge profiles via CEX-HPLC.
- Identify the cell line (and specific clone) that provides the greatest increase in the main peak and the greatest decrease in the acidic peak [68].
- Process Parameter Optimization:
- Use a DoE approach to define the best process parameters (e.g., pH, temperature shift timing, dissolved oxygen) for the selected cell line to maintain high productivity while ensuring the optimal charge profile [68].
Protocol 3: A Machine Learning Workflow for Media and Process Optimization
For complex, non-linear interactions between culture parameters, machine learning (ML) offers a powerful alternative to traditional DoE, potentially reducing experimental burden by 3- to 30-fold [70] [18].
Procedure
The following workflow diagrams the ML-driven optimization process, from data collection to model validation.
Key Steps
- Define Inputs and Outputs: Input factors (e.g., concentrations of media components, pH, temperature) and target CQAs (e.g., % main charge variant, titer) are defined [18].
- Generate Initial Dataset: An initial set of data is collected, either from historical records or a small-scale DoE.
- Model Training: A supervised ML regression model (e.g., Gaussian Process) is trained to predict the CQAs based on the input factors [70] [18].
- Iterative Optimization: A Bayesian Optimization algorithm uses the model to propose new experiments that balance exploring uncertain regions of the design space and exploiting known promising regions [70].
- Validation: The predicted optimal conditions are validated experimentally to confirm improved charge variant profiles and productivity.
Results and Data Presentation
Quantitative Outcomes of Optimization Strategies
The following table summarizes typical quantitative improvements achievable through the systematic application of the described protocols.
Table 2: Summary of Optimization Strategies and Reported Outcomes
| Optimization Strategy | Key Parameters Addressed | Reported Outcome | Experimental Burden |
|---|---|---|---|
| Additive & Media Screening [68] | Chemical additives, basal/feed media composition | Significant increase in main peak; reduction in acidic variants | Medium (Requires systematic screening) |
| Cell Line Screening [68] | CHO host cell line (e.g., CHO-K1, CHO-S) | Identification of cell lines with inherently superior charge profiles | High (Requires generation and screening of multiple clones) |
| Design of Experiments (DoE) [68] | Process parameters (pH, temp, feed timing) | Maintained target titer while achieving desired charge profile | Medium |
| Machine Learning/Bayesian Optimization [70] [18] | All of the above, including complex interactions | Identified conditions with improved outcomes vs. standard methods | Low (3x-30x fewer experiments than DoE) |
The Scientist's Toolkit: Essential Research Reagents
This table lists key reagents and their functions for conducting upstream optimization experiments focused on charge variants.
Table 3: Key Research Reagent Solutions for Upstream Optimization
| Reagent / Solution | Function / Application | Example |
|---|---|---|
| Chemically Defined Media | Serum-free, animal-origin-free base media providing essential nutrients; minimizes variability and risk of contamination [71]. | ActiCHO P, EX-CELL 325 PF CHO [71] |
| Feed Supplements | Concentrated nutrients fed during culture to sustain cell growth and productivity, and modulate product quality [71]. | Cell Boost 7a & 7b [71] |
| Selection Agents | Used for selecting and amplifying transfected cells with the gene of interest [72] [69]. | Methotrexate (MTX), Glutamine Synthetase (GS) system, Zeocin [72] [69] |
| Chemical Additives | Specific compounds used to directly modulate PTMs and shift charge variant profiles [68]. | Identified via platform screening (e.g., S-OptiCharge) [68] |
| Analytical Standards | For calibrating instruments and identifying specific glycoforms or charge variants in analytical assays [12]. | 2-AB labeled N-glycan standards [71] |
Optimizing the upstream process through strategic cell line selection and media additive formulation is a powerful approach to controlling charge variants in monoclonal antibodies. While traditional methods like additive screening and DoE remain effective, emerging technologies like Bayesian Optimization and Machine Learning present a paradigm shift, offering the potential to achieve superior results with a significantly reduced experimental burden. By integrating these protocols and strategies, scientists can design more efficient and robust upstream processes, ensuring the consistent production of high-quality biotherapeutics with the desired charge variant profile.
Systematic Platform Approaches (e.g., S-OptiCharge) for Charge Profile Modulation
Charge heterogeneity is an inevitable and critical quality attribute (CQA) of monoclonal antibody (mAb) therapies, arising from post-translational modifications (PTMs) during bioprocessing. Variations in protein charge profiles, resulting from modifications such as deamidation, oxidation, sialyation, and glycosylation, can directly impact an antibody's safety, efficacy, and overall consistency [73] [68]. For instance, deamidation and sialyation add negative charges, while glycation can mask positive charges, each potentially altering the therapeutic protein's behavior in vivo [68]. Effective control and modulation of these charge variants are therefore essential for ensuring the development of robust, high-quality biopharmaceuticals.
The S-OptiCharge platform, developed by Samsung Biologics, represents a systematic upstream process (USP) development approach designed to optimize protein charge variants while maintaining desired expression titers [68]. This platform employs a stepwise methodology that combines rigorous screening with design of experiments (DoE) to precisely tune charge profiles to meet target product specifications. By addressing charge variants early in development, the platform mitigates the risk of quality issues in later stages, which can be caused by process changes such as cell-line replacement [68]. This application note details the experimental protocols and data generated using the S-OptiCharge platform, providing a framework for researchers and drug development professionals to implement systematic charge variant control.
S-OptiCharge Platform Workflow and Signaling Pathways
The S-OptiCharge platform operates on a structured workflow that methodically identifies and optimizes key process parameters influencing charge variant distribution. The following diagram visualizes this systematic approach and the logical relationships between its core components.
Diagram 1: S-OptiCharge platform workflow.
The logical flow of the platform begins with additive screening to identify media supplements that positively influence charge profiles. This is followed by concurrent cell line screening and basal/feed media screening to find the optimal combination for producing the desired charge variant profile. The process culminates in a Design of Experiments (DoE) phase to define the best process parameters that maintain both high product titer and the optimized charge profile [73] [68].
The following second diagram illustrates the cause-and-effect relationships and the "signaling pathway" of how different input parameters and process conditions influence the critical quality attribute (CQA) of charge variation.
Diagram 2: Parameter impact on charge variants.
The "signaling pathway" demonstrates that inputs like cell line selection, media composition, and process parameters directly drive specific PTMs. These PTMs, in turn, directly alter the resulting charge variant profile by shifting the balance between acidic, main, and basic species [68]. The goal of the S-OptiCharge platform is to control these inputs to maximize the main peak and minimize undesirable acidic and basic variants.
Experimental Protocols for Charge Variant Modulation
Protocol A: Additive Screening for Charge Profile Improvement
Objective: To identify media additives that significantly improve the main peak proportion while reducing acidic variants in the charge variant profile [68].
Methodology:
- Cell Culture: Perform a 14-day cell culture process, adding basal media at the beginning and feed media on specified days (e.g., days 3, 5, and 7) [68].
- Additive Testing: Supplement the media with 10 different additive candidates, each containing specific vitamins and nutrients, alongside a negative control.
- Analysis: Harvest samples and analyze charge profiles using Cation-Exchange Chromatography (CEX). Quantify the percentage of acidic, main, and basic peaks.
Key Reagents:
- Basal and Feed Media: Pre-selected combinations.
- Additive Candidates: 10 distinct chemical supplements.
- Analysis Instrument: Cation-Exchange Liquid Chromatography (CEX) system.
Protocol B: Cell Line and Media Screening
Objective: To screen Chinese Hamster Ovary (CHO) cell lines and basal/feed media combinations to identify the optimal conditions for maximizing the main charge variant peak [68].
Methodology:
- Phase 1 - Basal Media & Cell Line Screening:
- Test 10 different basal media types.
- Use 4 different CHO cell lines.
- Analyze charge profiles via CEX to identify the cell line and basal media that yield the greatest increase in the main peak.
- Phase 2 - Feed Media Screening:
- Select the top 2 basal media from Phase 1.
- Test them with 9 different feed media.
- Use the top 2 cell lines identified from Phase 1.
- Analyze charge profiles to determine the combination that provides the greatest decrease in the acidic peak and increase in the main peak.
Key Reagents:
- Cell Lines: 4 CHO cell lines.
- Media: 10 basal media types, 9 feed media types.
- Analysis Instrument: Cation-Exchange Liquid Chromatography (CEX) system.
Protocol C: Design of Experiments (DoE) for Process Optimization
Objective: To define the optimal process parameters that maintain high productivity (titer) while preserving the desired charge profile achieved through prior screening [68].
Methodology:
- Input Selection: Identify key process parameters (e.g., pH, temperature, dissolved oxygen, feed timing) that may influence both titer and charge variants.
- Experimental Design: Construct a DoE matrix to systematically assess the effects of these parameters and their interactions on CQAs (charge variants) and titer.
- Execution and Analysis: Run the experiments as per the design and collect data on titer and charge profiles.
- Modeling and Optimization: Build statistical models to understand the relationship between inputs and outputs. Identify the design space where the charge profile is optimized and the titer is maintained at a comparable level to control conditions [68].
Data Presentation and Quantitative Analysis
Table 1: Summary of quantitative outcomes from systematic S-OptiCharge optimization steps.
| Optimization Step | Conditions Tested | Key Quantitative Outcome | Impact on Main Peak | Impact on Acidic Peak |
|---|---|---|---|---|
| Additive Screening [68] | 10 additives | 1 specific additive yielded a lower proportion of acidic and basic variants in one media combination. | Notably higher | Notably lower |
| Cell Line Screening [68] | 4 CHO cell lines, 10 basal media | 1 cell line provided the greatest increase in the main peak. | Greatest increase | - |
| Media Screening [68] | 2 basal media, 9 feed media, 2 cell lines | The same top cell line from initial screening showed a greater decrease in the acidic peak. | - | Greater decrease |
| DoE Optimization [68] | Multiple process parameters | Achieved a comparable titer to control conditions while maintaining the desired charge profile. | Maintained | Maintained |
Research Reagent Solutions and Essential Materials
Table 2: Key research reagents and materials used in the S-OptiCharge platform with their functions.
| Reagent / Material | Function in the Experimental Protocol |
|---|---|
| Chinese Hamster Ovary (CHO) Cell Lines | Host cells for expressing the recombinant monoclonal antibody; different clones can inherently produce different charge variant profiles [68]. |
| Basal Media | Provides initial nutrients and environment for cell growth and protein expression during the early stage of culture; its composition significantly impacts PTMs [68]. |
| Feed Media | Supplemented during the culture process to sustain cell viability and productivity; its composition is critical for modulating charge variants [68]. |
| Chemical Additives | Specific vitamins, nutrients, or chemicals added to media to directly influence charge profiles by altering the cellular environment or metabolic pathways [68]. |
| Cation-Exchange Chromatography (CEX) | The primary analytical technique used to separate and quantify the proportions of acidic, main, and basic charge variants in the protein sample [73] [68]. |
The S-OptiCharge platform provides a structured, data-driven framework for controlling a critical quality attribute in monoclonal antibody development. By employing a stepwise strategy of additive, cell line, and media screening, followed by statistical DoE optimization, it enables precise tuning of charge profiles early in the upstream process. This systematic approach ensures that target product quality is built into the process from the outset, mitigating downstream risks and contributing to the overall regulatory and clinical success of therapeutic proteins [68]. For researchers, adopting such a platform is instrumental in achieving a consistent, efficacious, and safe final biologic product.
Emerging Role of Machine Learning in Predicting and Controlling Charge Heterogeneity
Charge heterogeneity, a critical quality attribute (CQA) for monoclonal antibodies (mAbs), arises from post-translational modifications (PTMs) such as deamidation, glycosylation, and oxidation, forming acidic and basic variants that can significantly impact the stability, biological activity, and efficacy of therapeutic mAbs [18]. Regulatory agencies like the FDA and EMA require strict monitoring and control of charge variants during process development and optimization [18] [74].
Traditional optimization techniques, including one-factor-at-a-time (OFAT) and design of experiments (DOE), often fail to capture the complex, nonlinear interactions between culture parameters and medium components [18]. Machine learning (ML) has emerged as a powerful approach for modeling these intricate relationships, forecasting charge variant profiles, and enabling proactive control strategies within a Quality-by-Design (QbD) framework [18] [75]. This document details the application of ML for predicting and controlling charge heterogeneity, providing specific protocols and data analysis workflows for implementation in bioprocess development.
Machine Learning Approaches for Charge Variant Prediction
Machine learning models leverage large datasets from bioprocessing to uncover hidden patterns and predict the optimal conditions for minimizing undesirable charge variants, even when the underlying mechanisms are not fully understood [18].
Key Input Features and Predictive Targets
ML models are trained on historical data linking process parameters to final product quality. The table below summarizes common input features and the charge variant attributes they help predict.
Table 1: Key Input Variables and Output Targets for ML Models in Charge Heterogeneity Prediction
| Category | Specific Parameters | Impact on Charge Variants |
|---|---|---|
| Process Parameters | pH, temperature, dissolved oxygen (DO), culture duration [18] | High pH/temperature accelerate deamidation (acidic); low pH can promote basic variants [18]. |
| Media Components | Glucose, metal ions (e.g., Zn, Cu, Fe), amino acids, uridine [18] [76] | Fe and Zn significantly impact charge profile; uridine reduces acidic variants [76]. |
| Output Targets | Percentage of acidic variants, Percentage of basic variants, Main peak proportion [18] | Direct quantification of the charge heterogeneity profile, which is a CQA. |
Types of Machine Learning Models
Different ML algorithms are employed based on the nature of the data and the prediction goal.
- Supervised Learning and Regression: These methods are used to link process conditions with charge heterogeneity. Algorithms build a model based on labeled input data (process parameters) to predict continuous outputs (e.g., % acidic variants) [18].
- Explainable AI (XAI): Models like AbImmPred, built on the AntiBERTy language model, scan antibody sequences for immunogenic hotspots, providing insights beyond a simple prediction [75]. This is crucial for understanding model decisions and building scientific trust.
- Ensemble Modeling: This technique combines predictions from multiple algorithms to improve robustness and accuracy. For instance, holistic scoring systems integrate predictions for various biophysical properties into a unified developability score [75].
- Self-Driving Laboratories: These are AI systems that iteratively refine predictions using experimental feedback. Frameworks like SAMPLE and ProtAgents propose the future of autonomous antibody optimization, where AI agents design, test, and refine candidates based on key properties like charge heterogeneity [75].
Application Notes: ML-Driven Optimization Workflow
The following workflow and application notes outline a systematic approach for implementing ML in charge heterogeneity control.
The diagram below illustrates the integrated, cyclic process of data collection, model training, prediction, and experimental validation that forms the core of an ML-driven optimization strategy.
Case Study: Mitigating Acidic Variants with Fe and Zn Modulation
Background: An explainable AI study identified Fe and Zn as significant media components impacting the charge variant profile of a mAb [76]. The goal was to use ML to find their optimal concentrations to minimize acidic variants without compromising titer.
Data: Historical data from high-throughput experiments (e.g., 96-deepwell plate systems) exploring different concentrations of Fe, Zn, and other components, along with the resulting acidic variant percentage and titer, were used [76].
Model Training & Outcome: A regression model (e.g., Random Forest) was trained. The model successfully captured the non-linear relationship between metal ion levels and acidic variant formation. It predicted that a specific combination of reduced Fe and slightly increased Zn would lower acidic variants by ~15% while maintaining cell growth and productivity [76].
Validation: The predicted condition was validated in a lab-scale bioreactor. icIEF analysis confirmed a reduction in acidic variants from 25% to 21%, aligning with the model's prediction and demonstrating the practical utility of the ML-driven approach [76].
Experimental Protocols
This section provides detailed methodologies for the key experimental and computational procedures referenced in the application notes.
Protocol 1: Data Generation via High-Throughput Microscale Fed-Batch Cultures
This protocol generates the high-quality dataset required for training robust ML models [76].
1.0 Objective: To efficiently explore the impact of media and feed components on mAb charge heterogeneity using a high-throughput system.
2.0 Materials:
- CHO-S cell line expressing the mAb of interest.
- Basal media and feed concentrates.
- 96-deepwell plate system with gas-permeable seals.
- Microplate shaker incubator (e.g., INFORS HT Microtron).
- Metabolite analyzer (e.g., Nova Bioprofile).
- icIEF or CEX system for charge variant analysis.
3.0 Procedure:
- Experimental Design: Define a Design of Experiments (DoE) with factors such as pH, temperature, and concentrations of key components like Fe, Zn, and uridine.
- Inoculation: Inoculate each well of the 96-deepwell plate with a standardized cell density in a defined basal medium.
- Feeding Strategy: Apply feed solutions according to the DoE plan at specified timepoints (e.g., days 3, 5, 7).
- Process Monitoring: Sample daily to measure viable cell density, viability, and metabolite levels (e.g., glucose, lactate).
- Harvest: Centrifuge culture samples at the end of the run (e.g., day 14) to collect cell-free supernatant.
- Product Quality Analysis: Purify mAb from the supernatant using protein A affinity chromatography or analyze the supernatant directly [18]. Quantify charge variants using icIEF or CEX (see Protocol 2).
4.0 Data for ML: For each experimental run, compile a data vector containing all input process parameters and the corresponding output CQAs (e.g., % acidic, % basic, titer).
Protocol 2: Charge Heterogeneity Analysis by icIEF
Imaged capillary isoelectric focusing (icIEF) is the high-throughput gold-standard technique for charge variant analysis [42] [15].
1.0 Objective: To separate and quantify charge variants of a mAb based on their isoelectric point (pI).
2.0 Materials:
- icIEF instrument (e.g., Maurice, iCE3).
- Capillary cartridge.
- Pharmalyte 5-8 and 8-10.5.
- pl markers (e.g., 5.5, 7.0, 8.5, 10.0).
- Methylcellulose (0.35% w/v) or urea as stabilizer.
- Anolyte (e.g., 80 mM phosphoric acid) and catholyte (e.g., 100 mM sodium hydroxide).
- Master mix solution containing 4.5 M urea, 0.25% methylcellulose, 2.5% carrier ampholytes (Pharmalyte 5-8 and 8-10.5 in a 4:1 ratio), and 6.0 µg/mL peptide pI markers [15].
3.0 Procedure:
- Sample Preparation: Desalt the mAb sample. Mix the desalted mAb (final conc. ~0.5-1 mg/mL) with the master mix solution. Vortex and degas by centrifugation.
- Instrument Setup: Pre-focus the capillary with anode and cathode solutions at 1.5 kV for 1 minute.
- Loading and Focusing: Load the prepared sample mixture into the capillary. Focus the sample at 3.0 kV for 10 minutes.
- Imaging and Data Analysis: Image the focused zones using a CCD camera. The software will generate an electropherogram, identify peaks based on pI markers, and calculate the relative percentage of each charge variant (acidic, main, basic) based on peak area.
4.0 Notes: The method is highly reproducible and can be validated according to ICH Q2(R2) guidelines for use in quality control [42] [15].
Protocol 3: Implementing an ML Prediction and Control Model
This protocol outlines the computational steps for building a model to predict charge heterogeneity.
1.0 Objective: To develop a supervised ML model that predicts charge variant levels from process parameters and media composition.
2.0 Materials & Software:
- Dataset from Protocol 1 (CSV format).
- Python (v3.8+) with libraries: scikit-learn, pandas, numpy, matplotlib.
- Jupyter Notebook environment.
3.0 Procedure:
- Data Preprocessing:
- Load the dataset using
pandas. - Handle missing values (e.g., imputation or removal).
- Normalize or standardize numerical features (e.g., using
StandardScaler). - Split data into training (70%), validation (15%), and test (15%) sets.
- Load the dataset using
Model Training & Validation:
- Algorithm Selection: Test multiple algorithms (e.g., Random Forest, Gradient Boosting, Support Vector Machines).
- Hyperparameter Tuning: Use the validation set and grid search (
GridSearchCV) to find the optimal model parameters. - Cross-Validation: Perform k-fold cross-validation on the training set to assess model stability.
Model Evaluation:
- Predict charge variant percentages on the held-out test set.
- Calculate performance metrics: R², Mean Absolute Error (MAE), and Root Mean Squared Error (RMSE).
- A good model should have an R² > 0.8 on the test set.
Deployment for Prediction & Control:
- Use the trained model to predict outcomes for new, untested process conditions.
- Perform in-silico optimization (e.g., via differential evolution) to find parameter sets that minimize the objective function:
f(obj) = % Acidic Variants + penalty(Low Titer). - Validate the top predicted conditions experimentally (Protocol 1 and 2).
The Scientist's Toolkit: Key Research Reagent Solutions
The following table lists essential reagents and tools critical for conducting ML-driven charge heterogeneity studies.
Table 2: Essential Reagents and Tools for ML-Driven Charge Heterogeneity Research
| Item | Function/Description | Example Use Case |
|---|---|---|
| CHO Cell Culture Systems | Mammalian host for mAb production. | Generating process and quality data for ML training [18]. |
| DoE Software | Statistically plans efficient experiments. | Maximizing information gain from a limited number of bioreactor runs [18]. |
| icIEF Kit & Instrument | High-resolution charge variant analysis. | Quantifying acidic/main/basic species for model output/target [42] [15]. |
| Metal Ion Standards (Fe, Zn) | Controlled modulation of media components. | Investigating specific factors identified by ML as critical [76]. |
| Python with scikit-learn | Open-source platform for ML model development. | Building and validating regression models to predict charge variants [75]. |
| AI-Driven Developability Tools (e.g., AbImmPred) | Predicts immunogenicity from sequence. | Early-stage risk assessment of candidate mAbs [75]. |
Mitigating Common Artifacts and Challenges During Sample Preparation and Fractionation
Charge variant analysis is a critical quality attribute (CQA) assessment required by regulatory authorities for therapeutic monoclonal antibodies (mAbs) [36]. Charge heterogeneity arises from enzymatic or non-enzymatic post-translational modifications (PTMs) occurring during or after the biotechnological production process [43]. These modifications—including deamidation, glycation, oxidation, and C-terminal lysine variation—generate acidic and basic variants that can impact stability, potency, efficacy, and immunogenicity [2] [3]. A thorough characterization of these variants is essential for identifying CQAs and ensuring drug product quality [2].
The sample preparation and fractionation workflow presents multiple challenges where artifacts can be introduced, compromising analytical results and leading to inaccurate characterization. This application note details common artifacts, their mitigation strategies, and provides optimized protocols to support robust charge variant analysis in monoclonal antibody research and development.
Common Artifacts and Mitigation Strategies
The following table summarizes frequent artifacts encountered during sample preparation and fractionation for charge variant analysis, along with recommended solutions.
Table 1: Common Artifacts and Mitigation Strategies in Charge Variant Analysis
| Process Stage | Common Artifact | Impact on Analysis | Recommended Mitigation Strategy |
|---|---|---|---|
| General Sample Handling | Introduction of metal ions from stainless-steel surfaces [77] | Non-specific adsorption, peak tailing, low analyte recovery, metal-catalyzed oxidation [77] | Use bioinert column hardware (e.g., coated stainless-steel, PEEK-lined, titanium) and systems [77] |
| Buffer Preparation | Non-linear pH gradients [36] | Poor separation resolution, poor reproducibility between runs and operators [36] | Use commercially available pH gradient buffer kits designed to produce linear pH gradients [36] |
| Fraction Collection & Handling | Loss of labile modifications (e.g., succinimide) [3] | Incomplete characterization, inaccurate variant profiling [3] | Optimize sample preparation conditions (pH, temperature); avoid harsh denaturation [3] |
| Structural modifications during concentration, buffer exchange, or freeze/thaw [3] | Introduction of non-native variants, misidentification of degradation pathways [3] | Control and document fraction handling conditions; use side-by-side characterization of main peak as control [3] | |
| Scale-up to Semi-Preparative HPLC | Band broadening and poor peak resolution due to extra column volume [3] | Inability to isolate pure variants, cross-contamination between fractions [3] | Carefully optimize gradient methods for semi-preparative scale; ensure tubing lengths and components are appropriate [3] |
Experimental Protocols
Protocol 1: Semi-Preparative Fractionation of mAb Charge Variants
This protocol describes the isolation of charge variants using semi-preparative scale cation-exchange chromatography (CEX-HPLC) for subsequent characterization [3].
Materials and Equipment
- Monoclonal antibody sample (Drug Substance or Drug Product)
- Semi-preparative CEX column (e.g., with volatile salts for MS compatibility)
- HPLC system with fraction collector
- Mobile phase A (e.g., 25 mM ACES buffer, pH 7.0) [2]
- Mobile phase B (e.g., Mobile phase A with 150 mM Sodium Chloride) [2]
- Centrifugal concentrators with appropriate molecular weight cutoff
Procedure
- Sample Preparation: Dilute the mAb sample to 10 mg/mL using Mobile Phase A [2]. Centrifuge to clarify.
- Method Transfer & Optimization: Scale the analytical CEX method to the semi-preparative column. Adjust the gradient to account for differences in column dimensions and flow rates to maintain resolution [3].
- System Equilibration: Equilibrate the semi-preparative CEX column with 92% A / 7% B until a stable baseline is achieved [2].
- Sample Injection & Separation: Inject the sample. Elute charge variants using a optimized salt or pH gradient. Monitor the separation at 280 nm.
- Fraction Collection: Collect individual peak fractions based on UV signal. For low-abundance variants, pool corresponding fractions from multiple injections to obtain sufficient material [3].
- Purity Assessment: Analyze an aliquot of each collected fraction using the original analytical CEX method. Overlay the chromatogram with the unfractionated sample to confirm enrichment and purity (>80% enriched is target) [3].
- Sample Concentration & Buffer Exchange: If needed, concentrate fractions and exchange into a suitable storage buffer using centrifugal concentrators. Aliquot and store at -80°C.
Protocol 2: Enzymatic Treatment for Variant Confirmation
This protocol uses carboxypeptidase B (CPB) to remove C-terminal lysine, helping to confirm its contribution to basic variants [3].
Materials and Equipment
- Isolated basic variant fractions and main peak fraction (control)
- Carboxypeptidase B (CPB)
- Appropriate reaction buffer (e.g., neutral pH Tris or phosphate buffer)
- Thermostatic mixer
Procedure
- Sample Preparation: Transfer 50-100 µg of the isolated basic variant fraction and main peak control into separate vials. Adjust the buffer composition to be compatible with CPB activity if necessary.
- Enzyme Addition: Add CPB to the samples at a specified enzyme-to-substrate ratio. Include a control without enzyme.
- Incubation: Incubate the reaction mixture at 37°C for a predetermined time (e.g., 30-60 minutes).
- Reaction Termination: Stop the reaction by placing the samples on ice or by acidification.
- Analysis: Analyze the treated and untreated samples by analytical CEX. A shift in the peak's retention time towards the acidic region confirms the presence of C-terminal lysine in the original sample.
Workflow Visualization
The following diagram illustrates the key decision points and procedural steps in the fractionation workflow for characterizing mAb charge variants, incorporating critical mitigation strategies for artifacts.
Diagram 1: Fractionation Workflow with Mitigation Checkpoints
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Charge Variant Fractionation and Analysis
| Item | Function/Application | Key Considerations |
|---|---|---|
| Bioinert HPLC Columns [77] | Reduces non-specific adsorption of analytes to metal surfaces in column hardware. | Essential for maintaining recovery of electron-rich proteins and preventing metal-catalyzed modifications. Preferable to passivation. |
| Linear pH Gradient Buffer Kits [36] | Provides robust and reproducible linear pH gradients for CEX separation of charge variants. | Simplifies method development, improves consistency between labs and operators compared to in-house buffer preparations. |
| Cation Exchange Resins | High-resolution separation of mAb charge variants (acidic, main, basic species). | Select based on resolution requirements, capacity, and compatibility with MS (volatile salts). |
| Carboxypeptidase B (CPB) [3] | Enzymatic removal of C-terminal lysine residues under native conditions to confirm basic variant identity. | Must be used under conditions that do not denature the antibody or introduce other artifacts. |
| Sialidase [3] | Enzymatic removal of sialic acid residues to confirm contribution to acidic variants. | Helps identify and enrich other acidic variants that may co-elute with sialylated species. |
| IdeS Enzyme (FabRICATOR) [78] | Digests mAbs into Fc and F(ab')2 fragments at the hinge region for localized charge variant analysis. | Allows determination of whether charge differences originate from the Fab or Fc region of the antibody. |
Ensuring Product Quality: Functional and Comparative Analysis of Charge Variants
Within the framework of charge variant analysis in monoclonal antibody (mAb) research, the isolation of pure charge variants is a critical step for determining the structure-function relationship of these product-related species. Charge heterogeneity in mAbs arises from post-translational modifications such as deamidation, isomerization, C-terminal lysine processing, and oxidation [3]. These modifications can impact critical quality attributes including stability, potency, and serum half-life, potentially limiting product shelf-life and biological activity [3]. While analytical cation exchange chromatography (CEX) effectively monitors this heterogeneity, preparative CEX, particularly displacement chromatography, enables the isolation of individual variants for subsequent functional assessment. This application note details a protocol for isolating mAb charge variants using cation exchange displacement chromatography, providing researchers with a methodology to obtain highly enriched variants for comprehensive characterization.
Principles of Cation Exchange Displacement Chromatography
In displacement chromatography, separation occurs through a competitive adsorption process where a specialized displacer molecule with a higher affinity for the stationary phase than any sample component pushes the analytes down the column. The sample components compete for binding sites and are ultimately separated into consecutive displacement zones of pure substances that travel at the same velocity as the displacer front [79]. This method presents significant advantages for preparative separations:
- High Loading Capacity: The stationary phase is used more efficiently than in elution chromatography, allowing for larger quantities of material to be processed in a single run [79].
- Concentration Effect: Analytes are typically concentrated during the displacement process, reducing the need for post-isolation concentration steps.
- Overcoming Purity-Recovery Trade-offs: It is particularly effective for resolving closely related species, such as charge variants, where traditional linear gradient elution often results in a severe overlap between the main peak and variant peaks, creating a difficult trade-off between purity and recovery [79].
The following workflow diagram illustrates the key stages in the displacement chromatography process for charge variant isolation.
Materials and Reagents
Research Reagent Solutions
The successful implementation of this protocol requires the following key materials.
Table 1: Essential Research Reagents and Materials
| Item | Function | Examples & Specifications |
|---|---|---|
| Cation Exchange Resin | Stationary phase for separating charged mAb variants based on electrostatic interactions. | SP Sepharose Fast Flow, Monomix MC30 SP, or Nano SP-15L [79]. Select resin based on particle size (15-90 μm) and resolution requirements. |
| Carrier Buffer | Equilibrates the column and maintains a low ionic strength environment to promote tight binding of mAbs. | 20 mM Sodium Acetate, pH 5.0-5.5 [79] [46]. Buffer pH should be below the pI of the target mAb. |
| Displacer Solution | Competes with bound mAbs for cation exchange sites, driving the displacement process. | Protamine sulfate, dextran sulfate, or other high-affinity cationic polymers [79]. Concentration is critical and must be optimized. |
| Regeneration Buffer | Strips strongly bound substances, including the displacer, from the column to restore initial conditions. | 1-2 M Sodium Chloride in equilibration buffer. |
| Storage Buffer | Preserves column integrity during periods of non-use. | 20% Ethanol in water. |
| mAb Sample | The therapeutic protein containing the charge variants to be isolated. | Should be in a low-ionic-strength buffer compatible with the carrier buffer (e.g., dialyzed into 20 mM Sodium Acetate, pH 5.2) [79]. |
Equipment
- Chromatography System: ÄKTA or other FPLC/system with pumps, UV monitor, and fraction collector.
- Preparative Column: Appropriately sized for the required sample load.
- Analytical CEX Column: For assessing fraction purity (e.g., ProPac WCX-10, MAbPac SCX-10) [27].
- Buffer Preparation System: pH meter and filtration apparatus.
Experimental Protocol
Method Development and Optimization
Prior to preparative runs, initial scouting is essential to identify optimal conditions.
- Resin Screening: Evaluate different cation exchange resins for their resolution of the target mAb's charge variants. Smaller particle sizes (e.g., 15 μm) generally provide higher resolution compared to larger particles (e.g., 90 μm) [79].
- Displacer Selection and Titration: Test different displacers and a range of concentrations to find the condition that provides the best separation of the main mAb from its acidic and basic variants. An optimal displacer concentration is high enough to effect displacement but low enough to maintain sharp zone boundaries.
Table 2: Key Operational Parameters for Optimization
| Parameter | Typical Range | Influence on Separation |
|---|---|---|
| Buffer pH | 5.0 - 6.0 | Drives selectivity by altering the net charge of the mAb and its variants [79] [46]. |
| Sample Load | 10 - 50 mg/mL resin | Higher loading is possible in displacement mode; optimal load should be determined empirically. |
| Displacer Concentration | 0.1 - 10 mg/mL | Critical for sharp boundaries and resolution; must be optimized for each mAb-displacer pair. |
| Flow Rate | 0.1 - 0.5 mL/min (scale-dependent) | Shallower flow rates can improve mass transfer and resolution [79]. |
Step-by-Step Isolation Procedure
- Column Equilibration: Pack the selected cation exchange resin into a suitable preparative column. Equilibrate the column with at least 5 column volumes (CV) of the selected carrier buffer (e.g., 20 mM Sodium Acetate, pH 5.2) until the UV trace and pH of the effluent are stable.
- Sample Loading: Load the clarified and buffer-exchanged mAb sample onto the column. The sample should be in the same buffer as the carrier buffer to promote binding. High protein loading (e.g., 20-50 mg/mL resin) is typically applied to utilize the capacity of displacement chromatography [79].
- Washing: Wash the column with 3-5 CV of carrier buffer to remove any unbound or weakly bound proteins.
- Displacement: Initiate flow of the optimized displacer solution. Continue pumping the displacer until the UV signal indicates the emergence of the displaced component zones.
- Fraction Collection: Begin collecting fractions at the point where the UV signal first begins to increase significantly after the wash step. Continue collecting fractions throughout the elution of the displacement train.
- Regeneration and Storage: Once the displacement is complete, regenerate the column with 3-5 CV of high-salt regeneration buffer (e.g., 1 M NaCl) to remove any tightly bound materials, including the displacer. Re-equilibrate with carrier buffer or store in 20% ethanol.
Analysis of Isolated Fractions
Purity Assessment
- Analytical CEX Analysis: Analyze each collected fraction using an analytical CEX-HPLC method [3]. Overlay the chromatogram of each fraction with that of the unfractionated starting material.
- Acceptance Criterion: Isolated fractions should be highly enriched (>80% purity) for the target variant [3].
- Orthogonal Analysis: Confirm purity using an orthogonal technique such as Capillary Zone Electrophoresis (CZE) or imaged capillary isoelectric focusing (iCIEF) [5] [11].
Characterization and Functional Assessment
The following diagram outlines the characterization pathway for isolated fractions.
- Structural Identification:
- Employ intact mass analysis and peptide mapping with LC-MS to identify the specific chemical modification(s) responsible for the charge difference (e.g., deamidation, oxidation, glycation) [3].
- Use enzymatic treatment (e.g., Carboxypeptidase B to remove C-terminal lysine, sialidase to remove sialic acid) under native conditions to confirm the contribution of these modifications to the acidic or basic species [3].
- Functional Assays:
- Binding Assays: Perform Surface Plasmon Resonance (SPR) or ELISA to assess the impact of the variant on target antigen binding affinity and FcγR/FcRn binding [3].
- Cell-Based Assays: Evaluate the potency of the isolated variants using relevant cell-based bioassays to determine if the modification affects the mAb's mechanism of action (e.g., neutralization, antibody-dependent cellular cytotoxicity - ADCC) [3].
Discussion
Cation exchange displacement chromatography is a powerful tool for overcoming the classic purity-recovery trade-off encountered in the linear gradient elution of mAb charge variants [79]. By enabling high-loading and high-resolution isolation, it provides material of sufficient quantity and purity for definitive functional characterization. This is paramount for classifying variants as either product-related substances (comparable to the desired product) or product-related impurities (differing in activity, efficacy, or safety) per ICH Q6B guidelines [3]. Modifications in the Complementary Determining Region (CDR), such as deamidation, often reduce antigen binding affinity and are thus critical impurities to monitor and control [3]. The methodology outlined herein provides a robust framework for researchers to establish these critical structure-function relationships, ultimately ensuring the development of safe and efficacious biotherapeutic products.
Within the framework of charge variant analysis for monoclonal antibody (mAb) therapeutics, assessing functional potency is a critical regulatory requirement. mAbs are inherently heterogeneous, and post-translational modifications can lead to the formation of charge variants—commonly termed acidic and basic species—which have the potential to alter both antigen binding and Fc receptor interactions [37] [80]. While some studies indicate that the low-abundance charge variants typical of recombinant mAbs may not significantly impact potency or FcRn binding, the effects are ultimately dependent on the specific nature, location, and degree of the modifications [37]. This application note details standardized protocols for evaluating two critical quality attributes: antigen binding via a cell-based assay and FcRn binding affinity. These assays are essential for ensuring that charge variants do not adversely affect the biological functions and pharmacokinetic properties of antibody therapeutics.
Antigen Binding Assay: Simple Multiplexed Antibody-Binding Protocol
Background and Principle
Cell-based binding assays are a powerful tool for confirming that an antibody's mechanism of action, which often begins with binding to a cell surface receptor, remains intact despite charge heterogeneity [81]. The Simple Multiplexed Antibody-Binding Assay enables the simultaneous evaluation of antibody binding to target antigens presented on the surface of cells. A key strength of this protocol is the incorporation of internal negative controls within each well, which is achieved by using cell-permeant dyes to color-code positive and negative cell populations [82]. This allows for specific and reliable detection of binding, even when using crude supernatants.
Detailed Experimental Protocol
Materials & Reagents
- Antibody Sample: Purified mAb, hybridoma supernatant, or bacterial periplasmic extract.
- Cell Lines: A positive cell line expressing the target antigen and a negative control cell line that does not.
- Cell-Permeant Dyes: e.g., CFSE (for green fluorescence) and CellTracker Red (for red fluorescence), for cell color-coding.
- Staining Buffer: Phosphate-buffered saline (PBS) supplemented with 2-5% fetal bovine serum (FBS).
- Detection Antibody: A fluorescently-labeled secondary antibody specific to the Fc region of the test antibody (e.g., anti-human IgG-FITC).
- Equipment: Flow cytometer or fluorescence microscope, multiwell plates, centrifuge.
Procedure
- Cell Preparation and Staining:
- Harvest the positive and negative control cell lines using a gentle dissociation method.
- Resuspend each cell line in a separate tube with serum-free media containing a different cell-permeant dye. For example, stain the positive cell line with CFSE (green) and the negative cell line with CellTracker Red (red).
- Incubate the cells for 20-30 minutes at 37°C. Protect from light.
- Wash the cells twice with excess staining buffer to remove unbound dye.
- Combine the stained positive and negative cell populations into a single suspension.
Antibody Binding:
- Dispense the mixed cell suspension into a multiwell plate.
- Centrifuge the plate to pellet the cells and carefully remove the supernatant.
- Add the antibody sample (at various concentrations for a dose-response curve if needed) to the cell pellet. Resuspend the cells gently.
- Incubate for 60 minutes on ice or at 4°C to allow for binding while minimizing internalization.
Detection:
- Wash the cells twice with cold staining buffer to remove unbound antibody.
- Resuspend the cell pellet in a solution containing the fluorescently-labeled secondary antibody.
- Incubate for 30-45 minutes on ice, protected from light.
- Wash the cells twice with cold staining buffer to remove excess secondary antibody.
Analysis:
- Resuspend the final cell pellet in staining buffer and analyze by flow cytometry.
- Gate on the green (positive) and red (negative) cell populations separately.
- The mean fluorescence intensity (MFI) of the secondary antibody channel within the green (positive) cell population, after subtracting the signal from the red (negative) population, is proportional to the test antibody's binding to its target antigen.
Data Interpretation
A dose-response curve can be generated by plotting the MFI (or the percentage of bound cells) against the antibody concentration. The effective concentration (EC50) can be calculated to quantify binding affinity. A shift in the EC50 or a reduction in the maximum signal (efficacy) of a charge variant fraction compared to the main species may indicate a modification that compromises antigen binding [37].
FcRn Binding Affinity Assay
Background and Principle
The neonatal Fc receptor (FcRn) is pivotal in extending the half-life of IgG by recycling them from the endosome back to the bloodstream [83] [84]. This interaction is pH-dependent, with strong binding at acidic endosomal pH (~6.0) and minimal binding at neutral blood pH (~7.4) [84] [83]. Modifications that alter the surface charge of the Fc region can potentially disrupt this interaction, leading to accelerated clearance [37]. The HTRF FcRn Binding Assay is a robust, homogeneous method for quantifying this critical interaction [85].
Assay Principle: The assay is competitive. A biotinylated human FcRn ectodomain is bound to Streptavidin conjugated to a Terbium cryptate (Donor). A human IgG1 labeled with d2 (Acceptor) is used as the tracer. When the tracer antibody binds to FcRn, the Donor and Acceptor are brought close, and a FRET signal is emitted upon excitation. An unlabeled test antibody will compete with the d2-labeled tracer for binding to FcRn, resulting in a decrease in the FRET signal that is proportional to its concentration and affinity [85].
Detailed Experimental Protocol
Materials & Reagents
- HTRF FcRn Binding Kit: Includes biotinylated FcRn, Streptavidin-Tb cryptate, and d2-labeled IgG1 tracer [85].
- Antibody Samples: Main antibody species and pre-fractionated acidic/basic charge variants.
- Assay Buffer: Low pH buffer (e.g., pH 5.5-6.0) to mimic endosomal conditions.
- Equipment: A plate reader capable of measuring HTRF/TR-FRET signal, white or black multiwell plates.
Procedure
- Reagent Preparation:
- Reconstitute all kit components as per the manufacturer's instructions.
- Pre-incubate the biotinylated FcRn with Streptavidin-Tb cryptate to form the Donor complex.
Assay Setup:
- In a multiwell plate, first dispense the pre-formed Donor complex (FcRn-Streptavidin-Tb).
- Add the test antibody samples (main species and charge variants) at a range of concentrations. Include a control well with no competing antibody (maximum signal) and a blank well with no tracer (background signal).
- Add the d2-labeled IgG1 tracer to all wells.
- The sample volume is typically 10 µL per well after all additions [85].
Incubation and Reading:
- Incubate the plate for 2 hours at room temperature, protected from light.
- Read the plate using a TR-FRET-compatible plate reader. The excitation wavelength is typically 337 nm, and emissions are read at 620 nm (Donor) and 665 nm (Acceptor).
Data Calculation:
- The HTRF signal is calculated as the ratio of the emission at 665 nm over that at 620 nm, multiplied by 10,000.
- The background signal from the blank well is subtracted from all other wells.
- The signal in the presence of a competitor is expressed as a percentage of the maximum signal (no competitor control).
Data Interpretation
A standard competitive binding curve is generated by plotting the normalized HTRF signal against the logarithm of the competitor antibody concentration. The IC50 value (the concentration of competitor that inhibits 50% of the signal) is determined. A higher IC50 value for a charge variant compared to the main species indicates a lower affinity for FcRn, which could predict a reduced serum half-life.
Table 1: Example FcRn Binding Data for mAb Charge Variants
| Antibody Sample | IC50 (nM) | Relative Binding Affinity (% of Main Species) |
|---|---|---|
| Main Species | 45.2 | 100% |
| Acidic Species | 68.9 | 66% |
| Basic Species | 42.1 | 107% |
Visualizing Experimental Workflows and Signaling
Antigen Binding Assay Workflow
FcRn Binding Assay Principle
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for Antigen and FcRn Binding Evaluation
| Item | Function/Description | Example Application |
|---|---|---|
| Cell Permeant Dyes (e.g., CFSE, CellTracker) | Fluorescently label live cells for multiplexing and internal negative controls in cell-based binding assays [82]. | Simple Multiplexed Antibody-Binding Assay |
| Fluorescently-Labeled Secondary Antibodies | Detect the binding of a primary, unlabeled test antibody to cells for quantification by flow cytometry. | Antigen binding detection |
| HTRF FcRn Binding Kit | A complete reagent kit for homogeneous, competitive binding assays to quantify IgG-FcRn affinity at endosomal pH [85]. | FcRn affinity measurement |
| Lumit FcRn Binding Immunoassay | A homogenous no-wash competition assay based on NanoBiT technology to measure FcRn-Fc interactions [86]. | Alternative FcRn binding assay |
| Cation Exchange Chromatography Columns (e.g., ProPac SCX-10) | High-resolution separation of mAb charge variants (acidic/main/basic species) for subsequent functional analysis [36] [37]. | Fractionation of charge variants |
| pH Gradient Buffers | Generate linear pH gradients for robust, reproducible charge variant separation in cation exchange chromatography [36]. | Charge variant analysis |
Charge heterogeneity is a critical quality attribute of monoclonal antibody (mAb) therapeutics, arising from post-translational modifications (PTMs) such as deamidation, oxidation, C-terminal lysine processing, and glycation [19] [3]. These modifications can alter the isoelectric point (pI) of an antibody, potentially impacting its biological activity, stability, and pharmacokinetics (PK) [19] [12]. For biosimilar development, demonstrating comparability of charge variant profiles to the innovator product is a regulatory requirement [36] [12].
This application note details integrated strategies for isolating mAb charge variants and conducting comparative PK studies in animal models. We provide established protocols and a framework for data interpretation to assess the impact of charge heterogeneity on antibody clearance and exposure, which is essential for ensuring product quality and efficacy.
Charge Variant Isolation and Characterization
Isolation of Charge Variants
The first critical step is the separation and isolation of acidic, basic, and main charge species from the mAb starting material.
- Primary Separation Technique: Cation exchange displacement chromatography is a powerful and scalable method for this purpose. It has been successfully used to isolate gram quantities of charge variants for subsequent in vitro and in vivo studies [19].
- Analytical Monitoring: Analytical-scale cation exchange chromatography (CEX) is used to monitor the purity of isolated fractions. Isolated materials should achieve high purity (e.g., >94%) for reliable interpretation of PK studies [19].
- Alternative Techniques: Capillary zone electrophoresis (CZE) has emerged as a high-resolution technique for charge variant separation and is highly suitable for biosimilarity assessment due to its selectivity [12].
After isolation, a comprehensive characterization of the variants is necessary. Table 1 summarizes the key analytical techniques employed.
Table 1: Analytical Techniques for Characterizing Isolated Charge Variants
| Technique | Function | Key Information Obtained |
|---|---|---|
| Size Exclusion Chromatography (SEC) | Assess size-based heterogeneity and purity [19]. | Aggregate and fragment content; confirms isolation process does not artificially induce aggregation. |
| Mass Spectrometry (MS) | Identify chemical modifications [12] [3]. | Precise molecular weight mapping to identify modifications like deamidation, oxidation, and glycation. |
| Isoelectric Focusing (cIEF) | Determine isoelectric point (pI) [36]. | Confirms pI differences between acidic, basic, and main peak variants. |
| Cell-Based & Binding Assays | Evaluate functional properties [19] [3]. | In vitro potency, antigen-binding affinity, and FcRn binding affinity at neutral and acidic pH. |
Forced Degradation for Variant Enrichment
Low-abundance variants can be enriched through controlled stress studies to facilitate isolation and characterization. Common forced degradation conditions include [3]:
- Thermal Stress: Incubation at elevated temperatures to induce aggregation and fragmentation.
- pH Stress: Exposure to acidic or basic conditions to promote deamidation and isomerization.
- Oxidative Stress: Treatment with hydrogen peroxide to generate oxidized species.
In Vivo Pharmacokinetic Study Design
A robust animal study is crucial for evaluating the comparative PK of isolated charge variants.
Experimental Model and Dosing
- Animal Model: Sprague-Dawley rats are a well-established model for initial PK studies. Their physiology allows for the assessment of FcRn-mediated recycling pathways relevant to human PK [19].
- Route of Administration: Both intravenous (IV) and subcutaneous (SC) administration should be evaluated. IV administration provides direct insight into clearance and distribution, while SC administration assesses potential impacts of charge on absorption from the injection site [19].
- Dosing Regimen: A single dose of 10 mg/kg per variant has been successfully used in prior studies. Isolated variants (acidic, basic, main peak) and the starting material should be dosed in parallel to allow for direct comparison [19].
The following workflow diagram outlines the key stages of the charge variant PK study, from sample preparation to data analysis.
Sample Collection and Bioanalysis
- Blood Sampling: Serial blood samples should be collected over a period of 2-4 weeks to adequately characterize the distribution and elimination phases. A typical schedule includes pre-dose, and post-dose at 5 minutes, 4 hours, 8 hours, 1, 2, 4, 7, 14, 21, and 28 days [19].
- Bioanalytical Method: Use a specific and sensitive immunoassay (e.g., ELISA) capable of detecting the therapeutic mAb in the collected serum or plasma. The assay must be validated for the animal matrix used.
Pharmacokinetic Data Analysis
Non-compartmental analysis (NCA) is typically used to calculate standard PK parameters. The key parameters for comparison are summarized in Table 2.
Table 2: Key Pharmacokinetic Parameters for Comparative Analysis
| PK Parameter | Description | Interpretation in Charge Variant Studies |
|---|---|---|
| AUC0-∞ | Area Under the Curve from time zero to infinity. | Primary indicator of total systemic exposure. |
| Cmax | Maximum observed serum concentration. | Important for SC administration; reflects absorption. |
| CL | Total Systemic Clearance. | Direct measure of the body's efficiency in eliminating the mAb. |
| t½ | Terminal Elimination Half-Life. | Related to the FcRn-mediated recycling efficiency. |
| Vss | Volume of Distribution at Steady State. | Indicates the extent of tissue distribution. |
Case Study and Data Interpretation
A seminal study investigating a recombinant humanized IgG1 provides a benchmark for expected outcomes. In this study, charge variants were isolated and administered to rats [19].
- Similar In Vitro Properties: The isolated acidic, basic, and main peak variants showed comparable in vitro potency and FcRn binding affinity to the starting material [19].
- No Significant PK Differences: Following both IV and SC administration, the serum concentration-time profiles of the charge variants overlapped extensively. No statistically significant differences in AUC, clearance, or half-life were observed [19].
- Interpretation: This demonstrates that for this particular mAb, the natural charge heterogeneity resulting from common PTMs was not sufficient to alter FcRn binding or interaction with the vascular endothelium to a degree that impacted PK in rats.
It is important to note that these results are specific to the mAb studied and the magnitude of charge differences. Deliberate engineering of the Fv region to have a significantly higher positive charge has been shown to increase non-specific clearance in preclinical models [87]. The following diagram illustrates the FcRn-mediated recycling pathway, a key mechanism influencing IgG PK, and where charge interactions may play a role.
The Scientist's Toolkit
This section lists essential reagents and instrumentation for executing the protocols described in this application note.
Table 3: Essential Research Reagents and Solutions
| Item | Function / Application |
|---|---|
| Cation Exchange Resin | For the initial separation and isolation of charge variants at analytical and preparative scales [19] [27]. |
| pH Gradient Buffer Kits | Provide robust, linear pH gradients for CEX-based charge variant separation, reducing method development time [36]. |
| Formulation Buffer | To stabilize isolated variants for storage and in vivo dosing (e.g., 20 mM histidine acetate, 120 mM sucrose, 0.02% polysorbate 20, pH 6.0) [19]. |
| Carboxypeptidase B (CPB) | Enzyme used to remove C-terminal lysine residues, helping to characterize the contribution of this modification to basic variants [3]. |
| Sialidase | Enzyme used to remove sialic acid residues, helping to characterize the contribution of this modification to acidic variants [3]. |
| UHPLC System with Diode Array Detector | High-pressure liquid chromatography system for performing high-resolution, reproducible CEX separations [36] [27]. |
| CE-MS System (e.g., ZipChip) | Capillary electrophoresis-mass spectrometry platform for rapid charge variant analysis with integrated mass identification [88]. |
In the development of therapeutic monoclonal antibodies (mAbs), ensuring product quality, efficacy, and safety is paramount. Recombinant mAbs are inherently heterogeneous due to multiple post-translational modifications (PTMs) and degradation events that occur during manufacturing and storage [37] [3]. This heterogeneity manifests as charge variants, typically categorized as acidic species, main species, and basic species relative to the main peak when separated by charge-sensitive techniques like cation-exchange chromatography (CEX) [37]. According to ICH Q6B guidelines, understanding these variants is crucial; some are acceptable as product-related substances, while others that impact safety or efficacy must be classified and controlled as product-related impurities [3].
A critical task during biopharmaceutical development is to determine whether these charge variants exhibit different biological activities compared to the main species. Modifications in the complementarity-determining regions (CDRs) can alter antigen binding, while changes in the Fc region can affect effector functions or binding to the neonatal Fc receptor (FcRn), thereby influencing serum half-life [37] [3]. This case study provides a detailed protocol for isolating charge variants from a therapeutic mAb and conducting a comparative assessment of their key biological activities.
Background on Charge Variants
Origins and Definitions
Charge variants in monoclonal antibodies are primarily generated through a variety of chemical modifications. The main species is the antibody that elutes as the major peak on chromatograms and typically consists of antibodies with common PTMs like N-terminal pyroglutamate formation, C-terminal lysine processing, and glycosylation with neutral oligosaccharides [37].
- Acidic Species: These variants elute earlier than the main peak in CEX. Common modifications leading to acidic species include deamidation of asparagine residues, sialylation of glycans, glycation, and the presence of non-classical disulfide bonds or fragments [37] [26]. Deamidation, particularly in the CDR, is a major cause and can result in the formation of isoaspartic acid (isoAsp) or aspartic acid (Asp), introducing a negative charge and potentially altering conformation [37].
- Basic Species: These variants elute later than the main peak in CEX. They often arise from the presence of unprocessed C-terminal lysine, proline amidation, sequence variants, or oxidized species [37] [89] [26]. Isomerization of aspartate residues can also contribute to basic species [26].
Table 1: Common Modifications Found in Charge Variants and Their Potential Impact
| Variant Type | Common Modifications | Potential Impact on Biological Activity |
|---|---|---|
| Acidic Species | Deamidation (especially in CDR), Sialylation, Glycation, Fragmentation, Trisulfide bonds [37] [26] | ↓ Antigen-binding affinity, ↓ Potency (if in CDR) [37] [3] |
| Basic Species | Unprocessed C-terminal Lysine, C-terminal Proline Amidation, Oxidation, Succinimide formation [37] [89] [26] | Typically minimal impact on activity for C-terminal Lys; potential impact if modification is in CDR or FcRn binding region [37] [3] |
The Imperative for Functional Comparison
While some modifications like C-terminal lysine are generally considered benign, others, particularly those in the CDR, can be critical [3]. For instance, deamidation or isomerization in the CDR has been demonstrated to reduce antigen-binding affinity and biological potency, thereby categorizing those variants as product-related impurities [3]. Consequently, a thorough characterization that includes isolating variants and testing their biological activity is a regulatory expectation to establish the safety and efficacy of the biotherapeutic product [3] [26].
Materials and Equipment
Research Reagent Solutions
The following reagents and instruments are essential for executing the charge variant analysis and functional characterization.
Table 2: Key Research Reagent Solutions and Equipment
| Item Name | Function/Application |
|---|---|
| Cation-Exchange Column (e.g., Thermo Scientific ProPac WCX-10 or MAbPac SCX-10) [27] | High-resolution separation of mAb charge variants based on surface charge differences. |
| UHPLC System (e.g., Thermo Scientific Vanquish Flex UHPLC) [27] | Provides robust, high-performance liquid chromatography for precise variant separation. |
| pH Gradient Buffers (e.g., Thermo Scientific pH Gradient Buffer platform) [27] | Ready-to-use mobile phases for robust and reproducible pH gradient elution, saving method development time. |
| Chromatography Data System (CDS) Software (e.g., Thermo Scientific Chromeleon 7.3 CDS) [27] | For instrument control, data acquisition, and processing in compliant GxP environments. |
| Enzymes: Carboxypeptidase B (CpB), Sialidase [3] | Enzymatic treatment to remove C-terminal lysine (CpB) or sialic acid (Sialidase) for identifying contributions of these modifications. |
| Cell-Based Bioassay Reagents | To measure the potency and biological activity of isolated fractions. |
| Surface Plasmon Resonance (SPR) Instrument | For characterizing binding affinity (KD) to the target antigen and Fc receptors (e.g., FcγR, FcRn). |
Experimental Protocol
Charge Variant Separation and Fractionation
Principle: Utilize cation-exchange chromatography (CEX) with a pH gradient to separate mAb charge variants based on differences in their surface charge distribution [27] [46].
Procedure:
- Sample Preparation: Dilute the mAb drug substance to a concentration of 10 mg/mL using mobile phase A (e.g., 25 mM ACES buffer, pH 7.0) [26].
- Chromatographic System Setup:
- Column: Thermo Scientific MAbPac SCX-10 column (4 x 250 mm) or equivalent [27].
- Mobile Phase A: Low-pH buffer (e.g., 25 mM ACES, pH 7.0).
- Mobile Phase B: High-pH buffer (e.g., 25 mM ACES, 250 mM Sodium Chloride, pH 9.0). Volatile buffers can be used for subsequent mass spectrometric analysis [89].
- Gradient: Employ a shallow linear pH gradient from 0% to 100% mobile phase B over 10-30 minutes. The exact gradient may require optimization for the specific mAb [89].
- Flow Rate: 1.0 mL/min.
- Detection: UV absorbance at 280 nm.
- Injection Volume: 5-100 µL (semi-preparative scale) [26].
- Fraction Collection: Based on the UV chromatogram, collect fractions corresponding to the acidic region, the main peak, and the basic region into separate tubes. For higher purity, collect multiple sub-fractions across each peak [3].
- Fraction Processing: Concentrate the collected fractions using centrifugal filter units and perform buffer exchange into a neutral formulation buffer (e.g., PBS or histidine-sucrose buffer) suitable for downstream assays [3].
Figure 1: Workflow for charge variant separation and analysis.
Orthogonal Characterization of Isolated Fractions
Before functional testing, confirm the identity and purity of the isolated fractions.
- Purity Analysis: Re-analyze each fraction using the analytical CEX method. Overlay the chromatograms with the original, unfractionated sample to confirm enrichment and purity (>80% is desirable) [3].
- Intact Mass Analysis: Determine the molecular mass of the intact proteins in each fraction using LC-MS. This helps identify major modifications like glycation (+162 Da), lysine truncation (-128 Da), or oxidation (+16 Da) [89] [26].
- Peptide Mapping: For detailed characterization, digest the fractions with trypsin and analyze via LC-MS/MS. This pinpoints specific modification sites (e.g., deamidation at Asn 55 in CDR) and differentiates between isobaric species like Asp and isoAsp [37] [26].
Assessment of Biological Activity
Compare the functional properties of the acidic, basic, and main peak fractions using the following assays.
4.3.1 Antigen Binding Affinity Assay
- Method: Surface Plasmon Resonance (SPR) using a Biacore system.
- Protocol:
- Immobilize the target antigen on a CM5 sensor chip.
- Flow the isolated mAb fractions (at a series of concentrations, e.g., 0, 1.56, 3.125, 6.25, 12.5, 25, 50 nM) over the chip surface.
- Record the association and dissociation phases.
- Analyze the sensorgrams to calculate the binding kinetics: association rate (ka), dissociation rate (kd), and equilibrium dissociation constant (KD = kd/ka).
4.3.2 Fcγ Receptor Binding Assay
- Method: ELISA-based binding assay.
- Protocol:
- Coat a 96-well plate with recombinant FcγRIIIa (or other FcγRs).
- Block the plate with a protein-based blocking buffer.
- Add the isolated mAb fractions at a single concentration (e.g., 5 µg/mL) or a dilution series.
- Detect bound mAb using an enzyme-conjugated anti-human IgG antibody and a colorimetric substrate.
- Measure the absorbance and compare the signal between fractions.
4.3.3 Potency Bioassay
- Method: Cell-based assay relevant to the mAb's mechanism of action (e.g., ADCC reporter assay, neutralization assay).
- Protocol (Example ADCC Reporter Bioassay):
- Culture effector cells (e.g., engineered Jurkat cells expressing FcγRIIIa and a luciferase reporter under an NFAT response element) and target cells expressing the antigen.
- Co-culture the cells with the isolated mAb fractions in a dose-response manner.
- After incubation, add a luciferase substrate to the wells.
- Measure the luminescent signal, which is proportional to FcγR-mediated effector function.
- Calculate the relative potency (EC50) of each fraction compared to the main peak.
Data Analysis and Interpretation
Presentation of Quantitative Results
Data from the biological assays should be compiled into summary tables for direct comparison.
Table 3: Exemplary Data from Biological Activity Comparison of Isolated Charge Variants
| Sample Fraction | Antigen Binding KD (M) | Relative Antigen Binding (%) | FcγRIIIa Binding (Signal at 5 µg/mL) | Cell-Based Potency (Relative EC50 vs Main) |
|---|---|---|---|---|
| Main Peak | 1.25 x 10-9 | 100% | 1.45 | 1.00 |
| Acidic Peak | 2.50 x 10-9 | 50% | 1.42 | 0.95 |
| Basic Peak | 1.30 x 10-9 | 96% | 1.48 | 1.02 |
Interpretation and Correlation with Modifications
- Significant Reduction in Antigen Binding for Acidic Species: As shown in the exemplary data, a two-fold increase in KD (weaker affinity) and a 50% reduction in relative binding for the acidic fraction strongly suggests the presence of critical modifications within the Fab region, most commonly deamidation in the CDR [37] [3]. This finding would categorize this specific acidic species as a product-related impurity requiring tight control.
- Minimal Impact of Basic Species: The basic fraction shows binding and potency profiles nearly identical to the main peak. This is consistent with modifications like C-terminal lysine, which are located outside of binding regions and typically do not affect activity [3].
- Preserved Fc-Mediated Function: All fractions show similar FcγRIIIa binding and cell-based potency, indicating that the modifications present do not significantly impact the Fc effector function or the overall integrity of the mAb's mechanism of action in this assay format.
Figure 2: Relationship between PTM location and biological impact.
Discussion
The comprehensive characterization of charge variants is a cornerstone of biopharmaceutical development. As this case study demonstrates, a well-designed workflow from separation to functional analysis is critical for identifying critical quality attributes (CQAs). The data generated allows for science-based decisions regarding process development and control strategies to ensure a consistent and high-quality product.
A common challenge is the incomplete quantitative accounting of all acidic species, often leaving a "characterization gap" [26]. This can be due to difficulties in quantifying low-level, scattered modifications like glycation via peptide mapping, or the presence of conformational isomers that are separated by CEX but are not traditional PTMs [26]. Techniques like intact mass analysis for total glycation quantification and hydrogen-deuterium exchange mass spectrometry (HDX-MS) to probe higher-order structure should be employed if such gaps are identified [26].
The strategy outlined here—fractionation coupled with orthogonal analytical and functional techniques—provides a robust framework for understanding the structure-function relationship of mAb charge variants, ultimately ensuring the safety and efficacy of the therapeutic antibody.
Within the development of therapeutic monoclonal antibodies (mAbs), charge heterogeneity is a Critical Quality Attribute (CQA) that must be thoroughly characterized and controlled [2] [90]. Post-translational modifications (PTMs) occurring during production and storage, such as deamidation, oxidation, glycation, and C-terminal lysine variants, generate a spectrum of charge variants—commonly categorized as acidic, main, and basic species [2] [19] [3]. These variants can potentially impact the stability, biological activity, and efficacy of the drug product [12] [3]. Setting scientifically justified acceptance criteria for these variants is therefore a regulatory expectation, as outlined in guidelines such as ICH Q6B [36] [3]. This application note details a comprehensive, data-driven strategy for establishing these specifications, framed within the broader context of charge variant analysis research.
Experimental Design: A Multi-Technique Orthogonal Approach
A thorough characterization workflow is essential to link specific variants to potential impacts on product quality. The strategy involves separating, isolating, and intensively analyzing charge variants to identify their modifications and assess their biological relevance.
The following workflow outlines the core characterization process for identifying CQAs and setting specifications:
Charge Variant Separation and Isolation
The first critical step is the high-resolution separation of charge variants, typically using Ion-Exchange Chromatography (IEC) or imaged Capillary Isoelectric Focusing (icIEF) [2] [36] [90]. While icIEF is valued for its speed and reproducibility, preparative IEC is often used for its ability to isolate milligram quantities of variant fractions for downstream analysis [3]. A key challenge is scaling up analytical methods to semi-preparative scale while maintaining resolution, which requires careful optimization of column dimensions, flow rates, and gradients [3]. Collected fractions must be of high purity (>80% enriched) to ensure accurate characterization, which can be verified by re-injecting the fraction and confirming a single, sharp peak [3].
In-depth Characterization of Isolated Fractions
Once isolated, fractions are subjected to a suite of orthogonal analytical techniques. The table below summarizes the key methodologies and their specific roles in characterizing charge variants.
Table 1: Key Analytical Techniques for Charge Variant Characterization
| Technique | Primary Function | Key Information Obtained | Reference |
|---|---|---|---|
| Peptide Mapping with LC-MS/MS | Identify and localize specific PTMs | Exact sites and levels of deamidation, oxidation, glycation, etc. | [2] [12] |
| Intact Mass Analysis (LC-MS) | Determine total glycation and other mass changes | Global quantification of modifications like glycation (+162 Da) | [2] |
| Size-Exclusion Chromatography (SEC) | Monitor aggregation and fragmentation | Quantify high/low molecular weight species (HMW/LMW) | [2] [19] |
| Hydrogen-Deuterium Exchange MS (HDX-MS) | Probe higher-order structure (HOS) | Detect conformational changes not attributable to known PTMs | [2] |
| Cell-Based Bioassays / Binding Assays | Assess biological activity and potency | Determine functional impact of variants on antigen/FcRn binding | [2] [19] [3] |
| Differential Scanning Calorimetry (DSC) | Evaluate conformational stability | Measure thermal melting temperature (Tm) of domains | [91] |
Forced Degradation Studies
To enrich low-abundance variants and understand degradation pathways, forced degradation studies are employed [3]. Stressing the drug substance under conditions such as elevated temperature, extreme pH, light, or oxidants (e.g., hydrogen peroxide) can generate variants that are otherwise difficult to detect. These studies help establish the linkage between specific modifications and loss of product quality, providing a stronger scientific rationale for setting tight acceptance criteria for labile CQAs [3].
Data Integration and Setting Acceptance Criteria
The data from the above characterization workflow must be synthesized to make informed decisions on specifications. The following table provides a hypothetical example of how characterization data can be directly translated into acceptance criteria for a specific mAb.
Table 2: Example Data Integration for Setting Acceptance Criteria
| Charge Variant | Identified Modification(s) | Impact on In Vitro Potency | Impact on Pharmacokinetics (Rat) | Proposed Acceptance Criterion | Justification |
|---|---|---|---|---|---|
| Acidic Variants | Deamidation (CDR), Glycation, Fragments | Significant reduction if in CDR [3] | No significant impact observed [19] | NMT 25% | Control for variants with potency impact. |
| Basic Variants | C-terminal Lysine, Proline Amidation, Aggregates | No impact [19] | No significant impact [19] | NMT 15% | Control for process consistency and aggregates. |
| Main Peak | N/A | Reference | Reference | NLT 60% | Ensure majority species consistency. |
This data-driven approach moves beyond arbitrary limits. For instance, if characterization reveals that a specific deamidation in the CDR causes a loss of potency, the acceptance criterion for acidic variants must be set to control this specific impurity. In contrast, variants like C-terminal lysine, which show no impact on activity or pharmacokinetics in animal models [19], may be controlled to a less stringent level, primarily to ensure manufacturing consistency.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key reagents and instruments critical for successfully executing the charge variant characterization workflow.
Table 3: Essential Research Reagent Solutions for Charge Variant Analysis
| Item | Function / Application | Key Consideration |
|---|---|---|
| Cation-Exchange Column (e.g., ProPac SCX-10) | High-resolution separation of mAb charge variants using pH or salt gradients. | Enables direct MS coupling with volatile pH gradients [36]. |
| MS-Compatible Buffer Kits (pH gradient buffers) | Generate linear pH gradients for reproducible IEC separation. | Allows for robust and transferable methods across labs [36]. |
| IdeS / FabRICATOR Enzyme | Digests mAbs into Fc/2 and F(ab')2 fragments for domain-specific analysis. | Crucial for localizing modifications to specific antibody domains [91]. |
| Carboxypeptidase B (CPB) | Enzymatically removes C-terminal lysine from mAbs. | Used to confirm the identity of basic variants and simplify the charge variant profile [3]. |
| Sialidase | Removes sialic acid residues from glycans. | Used to confirm the contribution of sialylation to acidic variants [3]. |
| Reference Standard mAb (e.g., NISTmAb) | System suitability control and method benchmarking. | Ensures analytical method performance and inter-lab comparability [28]. |
Setting specifications for mAb charge variants is a fundamental activity in biopharmaceutical development that must be grounded in extensive analytical characterization. The integrated strategy outlined here—combining sophisticated separation techniques, orthogonal physicochemical and biological analyses, and forced degradation studies—provides the necessary evidence to define acceptance criteria that truly ensure drug product quality, safety, and efficacy. This rigorous, data-driven approach is indispensable for meeting regulatory standards and ultimately delivering safe and effective therapeutics to patients.
Demonstrating Comparability for Process Changes and Biosimilar Development
Within the development and lifecycle management of biotherapeutic monoclonal antibodies (mAbs), demonstrating comparability is a critical regulatory requirement. This process ensures that manufacturing process changes do not adversely impact the product's quality, safety, or efficacy [92] [93]. Similarly, for biosimilar development, a comprehensive comparative analytical assessment must establish that the biosimilar is highly similar to an originator reference product, notwithstanding minor differences in clinically inactive components [92] [94]. Charge variant analysis, mandated by guidelines such as ICH Q6B, serves as a cornerstone technique in these assessments, providing deep insight into the heterogeneity of therapeutic proteins caused by post-translational modifications (PTMs) and process-related changes [36] [3].
The inherent complexity and heterogeneity of mAbs necessitate robust analytical techniques for comparability exercises. Post-translational modifications such as deamidation, C-terminal lysine clipping, and glycosylation create a spectrum of charge variants that are critical quality attributes (CQAs) [3]. This application note details protocols for characterizing these charge variants, leveraging advanced chromatographic and electrophoretic techniques to support successful comparability and biosimilarity assessments.
Analytical Techniques for Charge Variant Analysis
Ion Exchange Chromatography with pH Gradients
Ion exchange chromatography (IEC) is a foundational technique for separating charge variants. While traditional salt-gradient IEC methods are effective, they often require extensive, molecule-specific method development and can lack reproducibility [36]. The use of pH gradients on cation exchange columns presents a more generic and robust alternative.
- Principle: Charge variants are separated based on their differential elution at a specific pH within a shallow, linearly increasing pH gradient. Variants with lower isoelectric points (pI) elute first, followed by the main species and then basic variants [36].
- Protocol - Fast pH-Gradient CEX-HPLC:
- Column: ProPac SCX-10, 4 x 250 mm (or equivalent).
- Mobile Phase A: 10 mM Sodium Phosphate buffer, pH 6.8.
- Mobile Phase B: 10 mM Sodium Phosphate buffer containing 500 mM Sodium Chloride, pH 10.5.
- Gradient: Linear pH gradient from 0% to 100% B over 30 minutes.
- Flow Rate: 1.0 mL/min.
- Detection: UV at 280 nm.
- Temperature: 25-30°C.
- Sample Load: 50-100 µg of mAb.
- Advantages: This method is globally applicable to mAbs with pI between 7-9, significantly shortens analysis time (30 min vs. 90 min for salt gradients), and offers superior reproducibility [36]. The use of commercially available pH gradient buffer kits can facilitate the generation of truly linear pH gradients, enhancing method robustness.
Capillary Zone Electrophoresis-Mass Spectrometry (CZE-MS)
Capillary zone electrophoresis (CZE) offers high-efficiency separation of charge variants and, when coupled with mass spectrometry (MS), enables direct identification of proteoforms.
- Principle: Charge variants are separated in a fused-silica capillary based on their electrophoretic mobility in a conductive background electrolyte (BGE). Direct coupling to MS allows for accurate mass determination of the resolved variants [5] [6].
- Protocol - MS-Compatible CZE-UV/MS:
- Capillary Coating: Successive multiple ionic-polymer layers (SMIL) based on diethylaminoethyl–dextran–poly(sodium styrene sulfonate) to generate a stable, cationic coating with controlled electroosmotic flow (EOF) [5]. Alternatively, a static neutral coating with hydroxypropyl methylcellulose (HPMC) can be used [6].
- Background Electrolyte (BGE): 50 mM volatile ammonium acetate buffer, pH 5.0 [6].
- Separation Voltage: +10 kV to -10 kV (polarity depends on coating).
- Injection: Hydrodynamic injection at 40 mbar for 5 seconds (using a 1 mg/mL mAb solution in BGE).
- MS Coupling: Utilize a low-flow sheath liquid interface (e.g., nanoCEasy) with a sheath liquid of 50% (v/v) acetonitrile with 0.1% formic acid at a flow rate of 0.8–1.0 µL/min.
- Advantages: CZE-MS provides high-resolution separation and direct characterization of variants like C-terminal lysine, sialic acid variants, and deamidation products. The use of volatile, MS-compatible BGEs is crucial for this application [5] [6].
Table 1: Comparison of Key Charge Variant Analysis Techniques
| Technique | Separation Mechanism | Key Application | Resolution | MS Compatibility |
|---|---|---|---|---|
| CEX-HPLC (pH Gradient) | Differential elution by pH | High-resolution profiling and preparative fractionation | High | Low (requires offline fractionation) |
| CZE-UV | Electrophoretic mobility in BGE | High-efficiency routine charge profiling | Very High | Poor with standard BGEs |
| CZE-MS | Electrophoretic mobility in volatile BGE | Identification and characterization of proteoforms | Very High | Excellent |
Fractionation for Detailed Characterization
For a comprehensive structure-function understanding, isolating charge variants for further analysis is often necessary.
- Protocol - Semi-Preparative Fraction Collection:
- Scale-Up: Transfer the analytical CEX-HPLC method to a semi-preparative scale column (e.g., 10 mm internal diameter) with careful gradient optimization to maintain resolution.
- Fractionation: Perform multiple injections to pool sufficient material (milligram level) for each variant peak. Collect acidic, main, and basic peaks individually.
- Purity Assessment: Re-analyze each collected fraction using the analytical CEX-HPLC method to ensure a purity of >80% enrichment.
- Downstream Analysis: Subject the purified fractions to orthogonal techniques, including peptide mapping with LC-MS/MS for PTM identification, glycan analysis, and cell-based bioassays to determine potency [3].
Experimental Data and Comparability Assessment
In a comparability study, the goal is to demonstrate that the pre-change and post-change products are highly similar. A robust charge variant analysis protocol is applied to both products, and the resulting profiles are quantitatively compared.
Table 2: Exemplary Quantitative Data from a Theoretical Comparability Study
| Charge Variant | Pre-Change Product (% Area) | Post-Change Product (% Area) | Acceptance Criterion | Status |
|---|---|---|---|---|
| Acidic Variants | 25.5% | 26.8% | NMT ± 5.0% | PASS |
| Sialylated Forms | 5.2% | 5.5% | Report | N/A |
| Deamidated Forms | 15.1% | 16.0% | Report | N/A |
| Main Peak | 68.0% | 67.0% | NMT ± 5.0% | PASS |
| Basic Variants | 6.5% | 6.2% | NMT ± 2.0% | PASS |
| C-terminal Lysine | 4.0% | 3.8% | Report | N/A |
The data in Table 2 shows that the relative proportions of charge variants in the post-change product fall within the pre-defined acceptance criteria, supporting a conclusion of analytical comparability.
Regulatory Framework and Strategic Considerations
Distinction: Comparability vs. Biosimilarity
A critical conceptual distinction exists between a comparability exercise and a biosimilarity assessment [92].
- Comparability: An assessment conducted by a single manufacturer to demonstrate that a manufacturing process change does not adversely impact the quality, safety, or efficacy of its own product. The comparison is between the pre-change and post-change product [92] [93].
- Biosimilarity: An extensive assessment conducted by a second manufacturer to demonstrate that its proposed biosimilar is highly similar to an originator's reference product, notwithstanding minor differences, and that there are no clinically meaningful differences in safety, purity, and potency [92] [94].
Post-Approval Changes for Biosimilars
The US FDA has clarified pathways for post-approval changes to licensed biosimilars. Changes (e.g., to formulation, dosage form, or strength) require a Prior Approval Supplement (PAS). The applicant must provide [95]:
- Comparability Data: Between the licensed biosimilar (pre-change) and the proposed product (post-change).
- Comparative Analytical Assessment (CAA) Data: This may involve re-using the original CAA data from the 351(k) BLA or conducting a new, targeted comparison between the proposed product and the reference product.
- Manufacturing Data: Such as process validation data.
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Charge Variant Analysis
| Item | Function/Description | Example Application |
|---|---|---|
| ProPac SCX-10 Column | A strong cation exchange column optimized for high-resolution separation of mAb charge variants using pH or salt gradients. | Separation of acidic and basic variants of denosumab and other mAbs [36]. |
| pH Gradient Buffer Kits | Commercially available buffer pairs designed to produce a truly linear pH gradient, enhancing method robustness and reproducibility. | Generic, fast (30 min) charge variant analysis for mAbs with pI 7-9 [36]. |
| Capillary Coating Reagents | Polymers (e.g., DEAE-Dextran, HPMC) used to coat fused-silica capillaries to control electroosmotic flow and prevent analyte adsorption. | Creating SMIL or neutral coatings for CZE and CZE-MS analyses [5] [6]. |
| Volatile BGE Salts | MS-compatible salts (e.g., Ammonium Acetate, Ammonium Formate) used to prepare background electrolytes for CZE-MS. | Enabling direct coupling of charge variant separation with mass spectrometric detection [6]. |
| Carboxypeptidase B (CPB) | Enzyme that specifically removes C-terminal lysine residues from antibodies under native conditions. | Enzymatic treatment to confirm the identity of basic variants and enrich other overlapping species [3]. |
| Sialidase | Enzyme that removes sialic acid residues from glycoproteins. | Enzymatic treatment to confirm the contribution of sialylation to acidic variants [3]. |
Experimental and Data Workflow Visualization
The following diagram illustrates the integrated workflow for charge variant analysis in a comparability study, from sample preparation to data interpretation.
Workflow for Comparability Assessment
A well-designed charge variant analysis strategy is indispensable for successful comparability and biosimilarity assessments. As demonstrated, techniques like pH-gradient CEX-HPLC and advanced CZE-MS provide the robust, high-resolution data required by regulators. By adhering to detailed experimental protocols and a structured workflow that includes fractionation and orthogonal testing, scientists can effectively monitor process changes and demonstrate the high similarity of biosimilar products, thereby ensuring the consistent quality, safety, and efficacy of biotherapeutic monoclonal antibodies.
Conclusion
Charge variant analysis is an indispensable pillar of mAb development, ensuring the production of safe, consistent, and efficacious biotherapeutics. A thorough understanding of the PTMs that cause heterogeneity, combined with a robust analytical toolbox for separation and characterization, allows scientists to identify Critical Quality Attributes. By implementing advanced troubleshooting and upstream optimization strategies, including emerging machine learning approaches, manufacturers can gain greater control over charge profiles. Ultimately, rigorous functional validation confirms that charge variants do not adversely impact biological activity or pharmacokinetics. The future of charge variant analysis lies in the deeper integration of advanced analytics, predictive modeling, and systematic process control to further streamline the development of next-generation antibody therapeutics.
References
- 1. Chromatographic analysis of the acidic and basic of... species [https://pubmed.ncbi.nlm.nih.gov/22820257/]
- 2. Challenges and Strategies for a Thorough Characterization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9687119/]
- 3. Characterization of Product Related Variants in ... [https://www.chromatographyonline.com/view/characterization-of-product-related-variants-in-therapeutic-mabs]
- 4. Controlling Charge During Variants ... Monoclonal Antibody [https://www.linkedin.com/pulse/controlling-charge-variants-during-monoclonal-mab-kashinath-mhatre-rutlc]
- 5. Charge variant analysis of monoclonal antibodies by CZE ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11952282/]
- 6. Characterization of monoclonal antibody charge variants ... [https://www.sciencedirect.com/science/article/pii/S0003267024010882]
- 7. Characterization of charge variants, including post- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10528315/]
- 8. Analytical Techniques for the Characterization and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9967501/]
- 9. Post-translational Modification in Antibody Function [https://www.antibodysociety.org/uncategorized/post-translational-modification-antibody-function/]
- 10. Characterization of charge variants, including post- ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914023008135]
- 11. Ion-exchange chromatography, capillary isoelectric ... [https://pubmed.ncbi.nlm.nih.gov/40741695/]
- 12. Biosimilarity and advanced structural characterization of ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025009889]
- 13. Identifying Post-Translational Modifications in Monoclonal ... [https://www.chromatographyonline.com/view/identifying-post-translational-modifications-monoclonal-antibodies]
- 14. Advancing Charge Variant Analysis with pH-Gradient Ion ... [https://www.waters.com/nextgen/nz/en/library/application-notes/2025/advancing-charge-variant-analysis-with-ph-gradient-ion-exchange-chromatography-optimizing-monoclonal-antibody-characterization-using-the-high-ph-kit-for-the-alliance-is-bio-hplc-system.html?srsltid=AfmBOopChdYEnGy6j3wj1bt_kiqWTitbiWlfkLqjaPa31_HU_afIXVoq]
- 15. Frontiers | Exploring imaged capillary isoelectric focusing parameters... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2025.1536222/full]
- 16. Oxidation and Deamidation of Monoclonal Antibody Products [https://pubmed.ncbi.nlm.nih.gov/34890632/]
- 17. Oxidation and Deamidation of Monoclonal Antibody Products [https://www.sciencedirect.com/science/article/abs/pii/S0022354921006468]
- 18. Machine learning-driven optimization of culture conditions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355708/]
- 19. Charge variants in IgG1: Isolation, characterization, in vitro ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3011216/]
- 20. of Recombinant Charge Variants Analysis Monoclonal Antibodies [https://biomedres.us/fulltexts/BJSTR.MS.ID.003395.php]
- 21. Small conformational changes in IgG1 detected as acidic ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269723002671]
- 22. Fc glycan sialylation of biotherapeutic monoclonal antibodies ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8564094/]
- 23. Characterization of the Charge Heterogeneity of a ... [https://www.mdpi.com/2073-4468/13/3/52]
- 24. Charge heterogeneity: Basic antibody charge variants with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5098448/]
- 25. C-terminal modification of monoclonal antibody drugs [https://www.sciencedirect.com/science/article/abs/pii/S0141813012003704]
- 26. Challenges and Strategies for a Thorough Characterization ... [https://www.mdpi.com/2306-5354/9/11/641]
- 27. Charge Variant Analysis | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/industrial/pharma-biopharma/biopharmaceutical-analytical-testing/intact-protein-analysis-workflows/charge-variant-analysis.html]
- 28. Ion-exchange chromatography, capillary isoelectric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12320836/]
- 29. Characterization of Biosimilar Monoclonal Antibodies ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12185665/]
- 30. Risk-Based Control Strategies of Recombinant Monoclonal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9703962/]
- 31. Charge-Variant Profiling of Biopharmaceuticals [https://www.chromatographyonline.com/view/charge-variant-profiling-biopharmaceuticals]
- 32. Charge Variant Analysis HPLC Columns [https://www.thermofisher.com/us/en/home/industrial/chromatography/liquid-chromatography-lc/hplc-uhplc-columns/biomolecule-hplc-uhplc-columns/charge-variant-analysis-hplc-columns.html]
- 33. Analyzing Charge Variant Profiles of Monoclonal ... [https://zenodo.org/records/15152615]
- 34. ICH Q6B Specifications: test procedures and acceptance ... [https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products-scientific-guideline]
- 35. ICH Q6B for Analytics [https://www.pharmtech.com/view/ich-q6b-for-analytics]
- 36. Charge Variant Analysis Information [https://www.thermofisher.com/us/en/home/industrial/pharma-biopharma/pharma-biopharma-learning-center/biopharmaceutical-characterization-information/aggregate-charge-variant-analysis-information.html]
- 37. Chromatographic analysis of the acidic and basic species ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3499298/]
- 38. Ion exchange chromatography of biotherapeutics [https://www.sciencedirect.com/science/article/pii/S0021967325000214]
- 39. Ion Exchange Chromatography in mAb Purification - Mabion [https://www.mabion.eu/science-hub/articles/ion-exchange-chromatography-in-monoclonal-antibodies-purification/]
- 40. Separation of Fab therapeutic charge variants by ion ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325003632]
- 41. Exploring imaged capillary isoelectric focusing parameters ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11904914/]
- 42. Development and Validation of an Optimized ... [https://www.sciencedirect.com/science/article/pii/S0928098725002957]
- 43. Fractionated charge variants of biosimilars: A review ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267021000015]
- 44. Structural and Functional Analysis of CEX Fractions Collected ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9415858/]
- 45. Native Anion Exchange Chromatography Coupled to Mass ... [https://www.chromatographyonline.com/view/native-anion-exchange-chromatography-coupled-to-mass-spectrometry-for-the-charge-variant-analysis-of-igg4-based-monoclonal-antibodies]
- 46. Protein concentration and analyzing charge variants in a ... [https://www.sciencedirect.com/science/article/abs/pii/S0378517324013723]
- 47. Advancing Charge Variant Analysis with pH-Gradient Ion- ... [https://www.waters.com/nextgen/si/en/library/application-notes/2025/advancing-charge-variant-analysis-with-ph-gradient-ion-exchange-chromatography-optimizing-monoclonal-antibody-characterization-using-the-high-ph-kit-for-the-alliance-is-bio-hplc-system.html?srsltid=AfmBOooXOZZ1XZ0_mH54I7RljLch0W5_XCFIk4sia2gpq7qBt-1CP55T]
- 48. Intact Mass Analysis of Antibodies [https://www.rapidnovor.com/services/antibody-protein-characterization/intact-mass-analysis/]
- 49. Peptide Mapping for Sequence Confirmation of ... [https://www.mdpi.com/1422-0067/26/20/9962]
- 50. Uncovering hidden protein modifications with native top ... [https://www.nature.com/articles/s41592-025-02846-5]
- 51. Tutorial: best practices and considerations for mass ... [https://www.nature.com/articles/s41596-021-00566-6]
- 52. Recommendations for Mass Spectrometry Data Quality ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3237091/]
- 53. CE-SDS Biologics Purity Analysis Kit [https://sciex.com/products/consumables/ce-sds-kit]
- 54. Systematic Development of a Size Exclusion ... [https://www.sciencedirect.com/science/article/pii/S0022354921001921]
- 55. Systematic Development of a Size Exclusion ... [https://pubmed.ncbi.nlm.nih.gov/33812889/]
- 56. Development and validation of capillary electrophoresis ... [https://www.sciencedirect.com/science/article/abs/pii/S0264410X23009428]
- 57. Development and validation of capillary electrophoresis ... [https://pubmed.ncbi.nlm.nih.gov/37591705/]
- 58. Targeted CQA analytical control strategy for commercial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11042060/]
- 59. mAb Therapies: Reducing Costs and Accelerating Timelines [https://www.bioprocessintl.com/monoclonal-antibodies/reducing-costs-and-accelerating-timelines-with-tools-for-physicochemical-characterization-of-mab-therapies]
- 60. Characterization of the acidic species of a monoclonal ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708519326603]
- 61. Charge variants characterization and release assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6512943/]
- 62. Forced degradation of recombinant monoclonal antibodies [https://pmc.ncbi.nlm.nih.gov/articles/PMC5680805/]
- 63. Forced Degradation Study as per ICH Guidelines [https://resolvemass.ca/forced-degradation-study-ich-guideline/]
- 64. Characterization of Product Related Variants in ... [https://www.chromatographyonline.com/view/characterization-of-product-related-variants-in-therapeutic-monoclonal-antibodies]
- 65. Assessing the Comparability of Degradation Profiles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12472202/]
- 66. The nonglycosylated variant in therapeutic monoclonal ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12341053/]
- 67. Optimization of Forced Degradation Using Experimental ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3097498/]
- 68. S-OptiCharge Platform Optimization of mAb Charge Profiles [https://www.bioprocessintl.com/sponsored-content/systematic-optimization-of-monoclonal-antibody-charge-profiles]
- 69. The Science of Cell Line Development for Biologics [https://www.patheon.com/us/en/insights-resources/blog/the-science-of-cell-line-development-for-biologics.html]
- 70. Accelerating cell culture media development using ... [https://www.nature.com/articles/s41467-025-61113-5]
- 71. Effect of different cell culture media on the production and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11928345/]
- 72. Optimized Split Zeocin Selection System Enhances ... [https://pubmed.ncbi.nlm.nih.gov/41211953/]
- 73. Improving therapeutic protein efficacy through charge profile adjustment | Samsung Biologics | The Leading Global CDMO with End-to-End Services [https://samsungbiologics.com/media/science-technology/improving-therapeutic-protein-efficacy-through-charge-profile-adjustment]
- 74. The Importance of Charge Heterogeneity in Monoclonal ... [https://www.linkedin.com/pulse/importance-charge-heterogeneity-monoclonal-antibody-kashinath-mhatre-w34ac]
- 75. AI-Powered Antibody Drug Development [https://www.genscript.com/learning-center/revolutionizing-developability-assessment-through-intelligent-innovation.html]
- 76. Machine learning-driven optimization of culture conditions ... [https://www.semanticscholar.org/paper/Machine-learning-driven-optimization-of-culture-and-Kavoni-Savizi/07cc897e6cb26b327fb6376a20600253a58a099c]
- 77. Solving Key Challenges in (Bio)pharmaceutical Analyses [https://www.chromatographyonline.com/view/solving-key-challenges-in-bio-pharmaceutical-analyses]
- 78. Characterization of charge variants of a monoclonal ... [https://www.sciencedirect.com/science/article/abs/pii/S0003269716304316]
- 79. Separation of monoclonal antibody charge variants using ... [https://www.sciencedirect.com/science/article/pii/S1383586619329776]
- 80. Molecular and functional analysis of monoclonal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5777976/]
- 81. In Vitro Potency Assays [https://pacificbiolabs.com/in-vitro-potency-assays/]
- 82. Simple Multiplexed Antibody-Binding Assay [https://pubmed.ncbi.nlm.nih.gov/29295900/]
- 83. The therapeutic age of the neonatal Fc receptor [https://www.nature.com/articles/s41577-022-00821-1]
- 84. Antigen physiochemical properties allosterically effect the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7531492/]
- 85. HTRF FcRn Binding kit, 100 Assay Points [https://www.revvity.com/product/htrf-fcrn-bind-kit-100-pts-64fcrnpet]
- 86. Lumit™ FcRn Binding Immunoassay Technical Manual [https://www.promega.com/resources/protocols/technical-manuals/500/lumit-fcrn-immunoassay-technical-manual-tm611/]
- 87. Evaluating the Use of Antibody Variable Region (Fv) ... [https://www.sciencedirect.com/science/article/pii/S0021925820394849]
- 88. Charge Variant Analysis [https://www.repligen.com/solutions/process-analytics/upstream/charge-variant-analysis]
- 89. Comprehensive characterisation of the heterogeneity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6343805/]
- 90. A new paradigm for optimized experimental design in cIEF ... [https://www.nature.com/articles/s41598-024-79108-5]
- 91. Comparison of enriched charge variants from different anti‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11926628/]
- 92. Differentiating biosimilarity and comparability in ... [https://pubmed.ncbi.nlm.nih.gov/27734233/]
- 93. Comparability Studies for Proteins and Biologics [https://www.intertek.com/pharmaceutical/biopharmaceuticals/comparability/]
- 94. Comparability studies and substitution of biosimilars [https://gabionline.net/biosimilars/research/Comparability-studies-and-substitution-of-biosimilars]
- 95. BioRationality: FDA Clarification Provides New Indications ... [https://www.centerforbiosimilars.com/view/biorationality-fda-clarification-provides-new-indications-and-process-change-for-biosimilars]