Complete Guide to Immune Cell Annotation with CellTypist: From Basics to Advanced Applications in Single-Cell RNA-Seq
This comprehensive guide provides researchers, scientists, and drug development professionals with essential knowledge for utilizing CellTypist in immune cell annotation of scRNA-seq data.
Complete Guide to Immune Cell Annotation with CellTypist: From Basics to Advanced Applications in Single-Cell RNA-Seq
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with essential knowledge for utilizing CellTypist in immune cell annotation of scRNA-seq data. Covering foundational concepts through advanced applications, the article explores CellTypist's logistic regression-based automated classification system, detailed methodological workflows for both built-in and custom models, optimization strategies for large datasets, and validation techniques against established cell ontologies. With practical examples from recent immunological studies and troubleshooting guidance, this resource enables accurate, reproducible cell type identification to accelerate research in immunology, disease mechanisms, and therapeutic development.
Understanding CellTypist: Core Principles and Immune Cell Annotation Fundamentals
What is CellTypist? Automated cell type annotation for scRNA-seq data
CellTypist is an automated cell type annotation tool specifically designed for single-cell RNA sequencing (scRNA-seq) data. It employs logistic regression classifiers optimised by a stochastic gradient descent algorithm to provide rapid and precise prediction of cell identities [1] [2]. Originally developed to explore tissue adaptation of immune cells, CellTypist has evolved into an open-source tool with a community-driven knowledge base for cell types, serving as a standardized platform for automated cell annotation [3]. One of its unique advantages is the comprehensive training set encompassing a wide range of immune cell types across diverse human tissues, enabling accurate organ-agnostic classification of immune compartments [2]. The tool is designed to recapitulate cell type structure and biology of independent datasets, providing robust models that are both scalable and flexible for integration into existing analysis pipelines [4].
Performance and Validation
Quantitative Performance Metrics
CellTypist has demonstrated high performance in cell type classification across multiple metrics. When trained on deeply curated and harmonized cell types from 20 different tissues across 19 reference datasets, CellTypist achieved precision, recall, and global F1-scores of approximately 0.9 for cell type classification at both high- and low-hierarchy levels [2]. The performance is notably robust to technical variations, including differences in gene expression sparseness between training and query datasets, as well as batch effects commonly encountered in scRNA-seq data [2].
Table 1: Performance Metrics of CellTypist Classifiers
| Classifier Hierarchy | Number of Cell Types | Precision | Recall | F1-Score |
|---|---|---|---|---|
| High-hierarchy (low-resolution) | 32 | ~0.9 | ~0.9 | ~0.9 |
| Low-hierarchy (high-resolution) | 91 | ~0.9 | ~0.9 | ~0.9 |
In comparative assessments with other label-transfer methods, CellTypist has shown comparable or better performance with minimal computational cost [2]. A notable advantage is its ability to resolve transcriptionally similar populations; for instance, it clearly distinguishes between monocytes and macrophages, which often form a transcriptomic continuum in scRNA-seq datasets due to their functional plasticity [2].
Comparison with Alternative Approaches
When benchmarked against emerging annotation methods, automated tools like CellTypist offer distinct advantages. Recent evaluations of GPT-4 for cell type annotation demonstrated its capability to generate expert-comparable annotations, with over 75% full or partial matches to manual annotations in most tissues [5]. However, CellTypist provides a specialized framework specifically optimized for scRNA-seq data analysis, avoiding potential limitations associated with large language models such as training corpus opacity and artificial intelligence hallucination risks [5].
Table 2: Comparison of Automated Cell Annotation Methods
| Method | Approach | Advantages | Limitations |
|---|---|---|---|
| CellTypist | Logistic regression with SGD | High performance (~0.9 F1-score), fast prediction, immune-focused | Organ-specific models may be needed for non-immune tissues |
| GPT-4 | Large language model | Broad knowledge base, no reference data needed | Undisclosed training corpus, potential hallucinations |
| SingleR | Correlation-based | Simple implementation, reference-based | Requires high-quality reference datasets |
| ScType | Marker-based | Marker gene focused, web application | Limited to predefined marker genes |
Installation and Setup
Installation Methods
CellTypist can be installed through multiple package management systems. For users with Python 3.6+ installed, the simplest approach is via pip:
Alternatively, installation through bioconda is also supported:
The installation includes dependencies such as pandas, scikit-learn, scanpy, and numpy, which are essential for the annotation workflow [6] [7].
Model Download and Configuration
CellTypist operates using pre-trained models that serve as the basis for cell type predictions. Users can download available models through the Python API:
The models are stored in a local directory (default: .celltypist/ in the user's home directory), though this path can be customized by setting the environment variable CELLTYPIST_FOLDER [1]. Since each model averages about 1 megabyte in size, downloading all available models is recommended for comprehensive analysis [1].
Core Annotation Workflow
Basic Annotation Procedure
The standard CellTypist workflow begins with importing the necessary modules and loading the query data. The input data should be a raw count matrix (reads or UMIs) in formats such as .txt, .csv, .tsv, .tab, .mtx or .mtx.gz, with cells as rows and gene symbols as columns [1]:
For data in gene-by-cell format, the transpose_input = True parameter should be specified. For MTX format files, additional gene_file and cell_file arguments are required to identify the feature and observation names [1].
Prediction Modes
CellTypist offers two distinct prediction modes to accommodate different annotation scenarios:
Best Match Mode (
mode = 'best match'): The default mode where each query cell is predicted to have the cell type with the largest score/probability among all possible types. This approach is straightforward and ideal for differentiating between highly homogeneous cell types [1].Probability Match Mode (
mode = 'prob match'): In this mode, a probability cutoff (default: 0.5, adjustable viap_thres) determines whether a cell is assigned to none, one, or multiple cell types. Cells failing the probability cutoff for all cell types receive an 'Unassigned' label, while those passing the cutoff for multiple types receive concatenated labels (e.g., "T cell|B cell") [1]. This mode is particularly valuable for identifying ambiguous cell states or novel cell types not well-represented in the reference model.
Majority Voting Refinement
To enhance annotation accuracy, CellTypist incorporates a majority voting approach that refines predictions within local cell clusters. When enabled (majority_voting = True), this feature performs over-clustering of the query dataset and assigns the dominant cell type label within each cluster [4] [8]. This strategy helps mitigate potential batch effects and improves consistency, as cells belonging to the same type are assigned identical labels regardless of technical variations [8].
The majority voting process generates additional columns in the output, including the original predictions, over-clustering assignments, and consensus labels after voting [9].
Output Interpretation and Visualization
Result Extraction
The AnnotationResult object returned by the annotate function contains three primary components:
predicted_labels: The main prediction results, including cell type assignments for each cell.decision_matrix: The raw decision scores for each cell across all cell types.probability_matrix: Probabilities transformed from the decision matrix using the sigmoid function [1].
These results can be exported to various formats for further analysis:
Visualization Methods
CellTypist provides built-in visualization capabilities to facilitate result interpretation:
The visualization function automatically generates UMAP coordinates using a canonical Scanpy pipeline, overlaying the predicted cell types for intuitive assessment of annotation quality [1].
Advanced Applications
Multi-Label Classification
For complex biological scenarios where cells may exhibit hybrid identities or transitional states, CellTypist supports multi-label classification. This approach is particularly valuable when dealing with unexpected cell types (e.g., low-quality cells or novel types) or ambiguous cell states (e.g., doublets) that fall outside the traditional "find-a-best-match" paradigm [6]. The multi-label capability allows CellTypist to assign zero (unassigned), one, or multiple cell type labels to each query cell, providing a more nuanced interpretation of cellular identities [6].
Custom Model Training
While CellTypist provides numerous pre-trained models, users can also train custom models on their own reference datasets:
The training process incorporates feature selection to identify the most informative genes for cell type discrimination, optimizing model performance and reducing computational requirements [9]. Custom models can be particularly valuable for specialized cell types or experimental conditions not adequately covered by the pre-trained models.
Online Interface
For users preferring a web-based approach, CellTypist offers an online interface accessible through the CellTypist portal [4] [8]. The online version accepts .csv or .h5ad files, with specific requirements for each format: CSV files should contain raw count matrices, while H5AD files require log-normalized expression data (normalized to 10,000 counts per cell) [8]. Results are delivered via email and include the same core components as the Python package: predicted labels, decision matrix, and probability matrix [8].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for CellTypist Workflow
| Tool/Resource | Function | Specifications | Application Context |
|---|---|---|---|
| CellTypist Python Package | Core annotation engine | Python 3.6+, requires scikit-learn, scanpy, pandas | Primary analysis tool for local execution |
| Pre-trained Models | Reference classifiers for prediction | ~1MB each; immune-focused (e.g., ImmuneAllLow.pkl) | Standardized cell type annotation without custom training |
| Raw Count Matrix | Input query data | Cells × genes (CSV, TSV, MTX, H5AD formats) | Essential input format for accurate prediction |
| Scanpy Ecosystem | Complementary analysis toolkit | Single-cell analysis pipeline for Python | Preprocessing, normalization, and visualization |
| CELLxGENE References | Curated data corpus | 22.2 million human cells, 164 cell types | Training data for model development and benchmarking |
Practical Implementation Protocol
Step-by-Step Experimental Guide
Data Preparation
- Format your scRNA-seq data as a raw count matrix with cells as rows and gene symbols as columns
- Ensure proper quality control has been performed (filtering of low-quality cells and genes)
- For log-normalized data (as needed for online interface), use
scanpy.pp.normalize_total(target_sum=1e4)followed byscanpy.pp.log1p()[8]
Model Selection
- For immune cell annotation: Start with
Immune_All_Low.pklorImmune_All_High.pkl[8] - Explore available models using
models.models_description()to identify tissue-specific options - Consider custom training if pre-trained models don't cover your cell types of interest
- For immune cell annotation: Start with
Annotation Execution
- Run basic annotation:
predictions = celltypist.annotate(input_file, model='selected_model.pkl') - Enable majority voting for cluster-refined results:
majority_voting=True - Use probability mode for complex populations:
mode='prob match', p_thres=0.5
- Run basic annotation:
Result Validation
- Examine confidence scores in the output (
insert_conf=True) - Compare with marker gene expression patterns
- Perform differential expression analysis between predicted clusters
- Utilize multi-label classifications to identify ambiguous populations
- Examine confidence scores in the output (
Downstream Analysis
- Integrate predictions with other analytical modalities (e.g., trajectory inference, gene regulatory networks)
- Compare CellTypist results with alternative annotation methods (e.g., manual annotation, other tools)
- Export results for publication-quality visualizations and further computational analysis
This comprehensive protocol ensures researchers can effectively implement CellTypist for their immune cell annotation research, from initial setup through advanced analytical applications.
The logistic regression classifier optimized by stochastic gradient descent
Algorithm Fundamentals
Logistic regression optimized by stochastic gradient descent (SGD) represents a powerful machine learning approach that combines the probabilistic interpretation of logistic regression with the computational efficiency of iterative gradient-based optimization. This method is particularly valuable in scenarios with large-scale datasets where traditional optimization methods become computationally prohibitive. The core concept involves applying a stochastic approximation of gradient descent to minimize the logistic loss function, resulting in faster iterations though with a potentially lower convergence rate compared to batch methods [10].
The fundamental objective function in logistic regression follows the form of a sum: Q(w) = 1/n * ΣQ_i(w), where w represents the parameters to be estimated, and each Q_i typically corresponds to the loss for an individual data point [10]. SGD optimizes this function by iteratively updating parameters using the gradient computed from individual samples or small mini-batches rather than the entire dataset, making it particularly suitable for large-scale problems in machine learning and statistical estimation [10].
Relevance to CellTypist in Immune Cell Annotation
Within the CellTypist ecosystem, logistic regression with SGD serves as the computational engine enabling rapid and accurate annotation of immune cell types from single-cell RNA sequencing (scRNA-seq) data [11] [2]. This implementation allows researchers to automatically transfer cell type labels from comprehensively curated reference models to query datasets, dramatically accelerating the analysis pipeline while maintaining biological accuracy [4]. The choice of SGD optimization is particularly strategic given the substantial sizes of modern scRNA-seq datasets, which frequently encompass hundreds of thousands of cells across numerous samples and conditions [2].
Algorithm Specification and Mathematical Formulation
Core Algorithm Components
The logistic regression classifier with SGD optimization integrates several mathematical components to achieve efficient model training:
Sigmoid Function: Transforms linear combinations of input features into probability estimates ranging between 0 and 1, representing the probability that a given sample belongs to a particular class [12].
Log Loss Function: Also known as cross-entropy loss, this function measures the discrepancy between predicted probabilities and actual class labels. The loss for a single sample is given by -1 * log(likelihood function), with the total loss representing the sum across all training samples [12].
L2 Regularization: Incorporated to prevent overfitting by penalizing large parameter values, enhancing model generalization to unseen data [12] [13]. The regularization strength is controlled through the parameter α (SGD) or C (traditional logistic regression), where C represents the inverse of regularization strength [13].
Parameter Update Mechanism
The SGD algorithm updates model parameters according to the following iterative process:
- Initialization: Initialize parameter vector
wand learning rateη[10] - Iteration Loop: Repeat until convergence criteria met [10]
- Data Shuffling: Randomly shuffle samples in training set [10]
- Sequential Processing: For each sample
i = 1,2,...,n, update parameters:w := w - η∇Q_i(w)[10]
For linear regression with a squared error loss, the parameter update takes the specific form:
This illustrates how each parameter is adjusted proportionally to the negative gradient of the loss with respect to that parameter [10].
Mini-Batch Extension
CellTypist implements an enhanced variant of SGD utilizing mini-batch training, where small batches of cells (typically 1,000 cells per batch) are processed sequentially rather than individual samples [14] [13]. This approach represents a compromise between computing the true gradient (using all data) and the gradient at a single sample, enabling better computational efficiency through vectorization while maintaining the beneficial stochastic properties of the algorithm [10].
Table 1: Comparison of Optimization Approaches in CellTypist
| Aspect | Traditional Logistic Regression | SGD Logistic Classifier | Mini-batch SGD |
|---|---|---|---|
| Data Usage | Entire dataset per iteration | Single random point per iteration | 1,000 cells per batch |
| Regularization | L2 with parameter C |
L2 with parameter α |
L2 with parameter α |
| Computational Efficiency | Lower for large datasets | Higher for large datasets | Highest for very large datasets |
| Convergence Behavior | Stable but slow | Noisy but fast | Balanced stability/speed |
| CellTypist Application | Small to medium datasets | Large datasets (>50k cells) | Very large datasets (>100k cells) |
Implementation Protocols for Immune Cell Annotation
CellTypist Model Training Procedure
The following protocol outlines the comprehensive procedure for training logistic regression models with SGD optimization using CellTypist for immune cell annotation:
Input Data Preparation:
- Format input data in cell-by-gene matrix structure (transpose if gene-by-cell) [13]
- Ensure expression data is log1p normalized to 10,000 counts per cell [13]
- Provide cell type labels as a list-like object or column name in AnnData metadata [13]
- Include gene identifiers corresponding to matrix columns [13]
Parameter Configuration:
- Set
use_SGD = Trueto enable stochastic gradient descent learning [13] - For large datasets (>100k cells), enable
mini_batch = Truefor enhanced efficiency [13] - Configure
batch_size = 1000andbatch_number = 100as default mini-batch parameters [13] - Set
epochs = 10as the default training iteration count [13] - Specify L2 regularization strength using
alpha = 0.0001(default) [13] - For traditional logistic regression (non-SGD), use
C = 1.0as inverse regularization strength [13]
Model Training Execution:
Model Validation and Application:
- Evaluate model performance using precision, recall, and F1-score metrics [2]
- Apply trained model to query datasets for cell type prediction [4]
- Optionally employ majority voting to refine predictions based on transcriptional similarity [15]
Advanced Training Configurations
For specialized applications, CellTypist offers several advanced training options:
Feature Selection Optimization:
- Enable
feature_selection = Truefor two-pass data training [13] - Set
top_genes = 300to select top genes from each cell type based on absolute regression coefficients [13] - Final feature set represents the union across all cell types [13]
Class Imbalance Adjustment:
- Activate
balance_cell_type = Trueto address imbalanced cell type frequencies [13] - Systematically oversamples rare cell types with higher probability [13]
- Ensures close-to-even cell type distributions in mini-batches [13]
Computational Performance Tuning:
- Specify
n_jobs = -1to utilize all available CPUs [13] - For traditional logistic regression, consider
use_GPU = Trueto enable GPU acceleration [13] - Adjust
max_iterbased on dataset size: 200 (large), 500 (medium), 1000 (small datasets) [13]
Table 2: CellTypist Training Parameters for Different Data Scenarios
| Scenario | use_SGD | mini_batch | batch_size | epochs | balancecelltype | feature_selection |
|---|---|---|---|---|---|---|
| Small dataset (<50k cells) | Optional | False | N/A | N/A | Optional | Recommended |
| Standard dataset (50-100k cells) | True | False | N/A | 10 | Optional | Recommended |
| Large dataset (100-500k cells) | True | True | 1000 | 10 | Recommended | Optional |
| Very large dataset (>500k cells) | True | True | 1000 | 10-30 | Highly Recommended | Optional |
| Imbalanced cell types | True | True | 1000 | 10-30 | True | Optional |
| High-dimensional data | True | Optional | 1000 | 10 | Optional | True |
Workflow Visualization and Experimental Design
CellTypist SGD Training Workflow
The following diagram illustrates the complete workflow for training logistic regression models with SGD optimization in CellTypist:
Immune Cell Annotation Pipeline
The comprehensive CellTypist annotation pipeline incorporating SGD-optimized logistic regression:
Performance Metrics and Validation
Model Evaluation Framework
CellTypist's logistic regression with SGD has been rigorously validated using comprehensive metrics and benchmarking:
Performance Metrics:
- Precision: Measures the accuracy of positive predictions [2]
- Recall: Quantifies the ability to identify all relevant instances [2]
- F1-Score: Harmonic mean of precision and recall, providing balanced assessment [2]
- Global F1-Score: Overall performance metric reaching approximately 0.9 for cell type classification [2]
Cross-Validation:
- Model performance remains robust against variations in gene expression sparseness [2]
- Prediction accuracy maintained despite batch effects between datasets [2]
- Performance correlates with representation of cell types in training data [2]
Table 3: CellTypist Performance on Immune Cell Annotation
| Evaluation Aspect | High-Hierarchy (32 types) | Low-Hierarchy (91 types) | Validation Method |
|---|---|---|---|
| Precision | ~0.9 | ~0.9 | Cross-dataset validation |
| Recall | ~0.9 | ~0.9 | Cross-dataset validation |
| F1-Score | ~0.9 | ~0.9 | Cross-dataset validation |
| Cell Types Identified | 15 major populations | 43 specific subtypes | Multi-tissue dataset |
| Robustness to Sparseness | High | High | Systematic testing |
| Batch Effect Resistance | High | High | Multi-dataset analysis |
| Computational Efficiency | High | High | Comparison to alternatives |
Comparative Performance Analysis
In benchmark studies, CellTypist demonstrated comparable or superior performance relative to other label-transfer methods while requiring minimal computational resources [2]. The tool successfully recapitulated immune cell biology across independent datasets, accurately resolving transcriptionally similar cell populations including:
- T cell heterogeneity (αβ vs. γδ T cells, CD4+ vs. CD8+ subsets) [2]
- B cell compartment (naive vs. memory B cells) [2]
- Mononuclear phagocytes (monocytes, macrophages, dendritic cells) [2]
- Innate lymphoid cells (NK cells, non-conventional T cells) [2]
- Dendritic cell subsets (DC1, DC2, migratory DCs) [2]
The granularity of annotation enabled the identification of tissue-specific immune features, such as distinct macrophage subpopulations in lung tissue characterized by expression of GPNMB, TREM2, and TNIP3 [2].
Research Reagent Solutions
Table 4: Essential Research Reagents and Computational Resources for CellTypist Implementation
| Resource Type | Specific Solution | Function/Purpose | Implementation Example |
|---|---|---|---|
| Data Input Format | Cell-by-gene matrix | Standardized input structure | AnnData objects, CSV/TSV/MTX files |
| Reference Data | Multi-tissue immune atlas | Training data for model development | 357,211 immune cells from 16 tissues |
| Preprocessing Tool | Scanpy pipeline | Data normalization and QC | Log1p normalization, HVG selection |
| SGD Implementation | Scikit-learn SGDClassifier | Core optimization algorithm | SGDClassifier(loss='log_loss') |
| Model Training | CellTypist.train function | End-to-end model training | celltypist.train(use_SGD=True) |
| Visualization | CellTypist.dotplot | Result visualization and interpretation | celltypist.dotplot(predictions) |
| Cluster Analysis | Leiden algorithm | Over-clustering for majority voting | scanpy.tl.leiden() integration |
| Performance Metrics | Precision/Recall/F1 | Model validation and benchmarking | Cross-dataset performance assessment |
| Gene Selection | Top_genes parameter | Feature selection for improved performance | Select 300 genes per cell type |
| Batch Correction | Built-in standardization | Handling technical variability | Data scaling during prediction |
Technical Notes and Troubleshooting
Optimal Parameter Selection
Based on extensive testing with immune cell datasets, the following guidelines ensure optimal performance:
Learning Rate Considerations:
- SGD requires careful learning rate tuning to balance convergence speed and stability [16]
- Excessive learning rates cause divergence, while insufficient rates slow convergence [10]
- CellTypist implements appropriate default values based on dataset characteristics [13]
Data Scaling Requirements:
- SGD performance is sensitive to feature scaling [16]
- Input data should be standardized for optimal results [16]
- CellTypist automatically performs appropriate scaling during training [13]
Regularization Strategy:
- L2 regularization prevents overfitting by penalizing large coefficients [12]
- Regularization strength (
αfor SGD,Cfor traditional) requires tuning based on dataset size and complexity [13] - Smaller
Cvalues (stronger regularization) may improve generalization at potential accuracy cost [13]
Troubleshooting Common Issues
Non-Convergence Solutions:
- Increase
max_iterparameter if cost function fails to converge [13] - Reduce learning rate for more stable convergence [10]
- Ensure proper data preprocessing and normalization [13]
Performance Optimization:
- For datasets >100,000 cells, enable
mini_batch=Truefor training efficiency [13] - Utilize
n_jobs=-1to parallelize computation across all available CPUs [13] - Consider feature selection (
feature_selection=True) for high-dimensional data [13]
Biological Validation:
- Always verify automated annotations with marker gene expression [2]
- Employ majority voting to refine predictions based on transcriptional similarity [15]
- Cross-reference identified populations with established immune cell signatures [2]
Within the field of single-cell RNA sequencing (scRNA-seq) analysis, accurate cell type annotation is a critical step for interpreting data and drawing meaningful biological conclusions. CellTypist has emerged as an automated tool that leverages logistic regression classifiers optimized by stochastic gradient descent to address this need [14] [4]. For researchers focusing on immune cells, the choice between the two primary built-in models, Immune_All_Low.pkl and Immune_All_High.pkl, forms a fundamental decision point that balances resolution against broad classification. These models are part of a curated collection available on the CellTypist website, where they can be downloaded for use within a Python environment [14] [1]. The "Low" and "High" suffixes refer directly to the hierarchy level of the cell types they contain; "Low" indicates low-hierarchy (high-resolution) cell types and subtypes, whereas "High" indicates high-hierarchy (low-resolution) cell types [14]. This protocol outlines a structured approach to exploring these models, enabling researchers to select the appropriate tool based on their experimental goals, whether for discovering novel immune subsets or for broader immune population mapping.
Model Characteristics and Quantitative Comparison
The Immune_All_Low and Immune_All_High models serve distinct purposes, and their differences are quantified in the table below. This comparison is essential for making an informed selection.
Table 1: Quantitative Comparison of CellTypist's Key Immune Models
| Feature | ImmuneAllLow | ImmuneAllHigh |
|---|---|---|
| Hierarchy Level | Low-hierarchy (High-resolution) | High-hierarchy (Low-resolution) |
| Number of Cell Types | 98 | 32 |
| Use Case | Detailed annotation of immune cell subtypes | Broad classification of major immune lineages |
| Example Annotation | Follicular B cells, Germinal center B cells, Memory B cells, Naive B cells [17] | B cells [17] |
| Recommended For | In-depth investigation of heterogeneous populations, novel subtype discovery | Initial data exploration, projects focused on major immune cell categories |
These models are built on a logistic regression framework, and for large training datasets, an SGD logistic regression approach using mini-batch training (e.g., 1,000 cells per batch) may be employed to enhance efficiency [14]. The models are serialized in a pickle format and can be easily inspected within Python to list all contained cell types and genes, providing transparency into the annotation process [1].
Experimental Protocols for Model Application
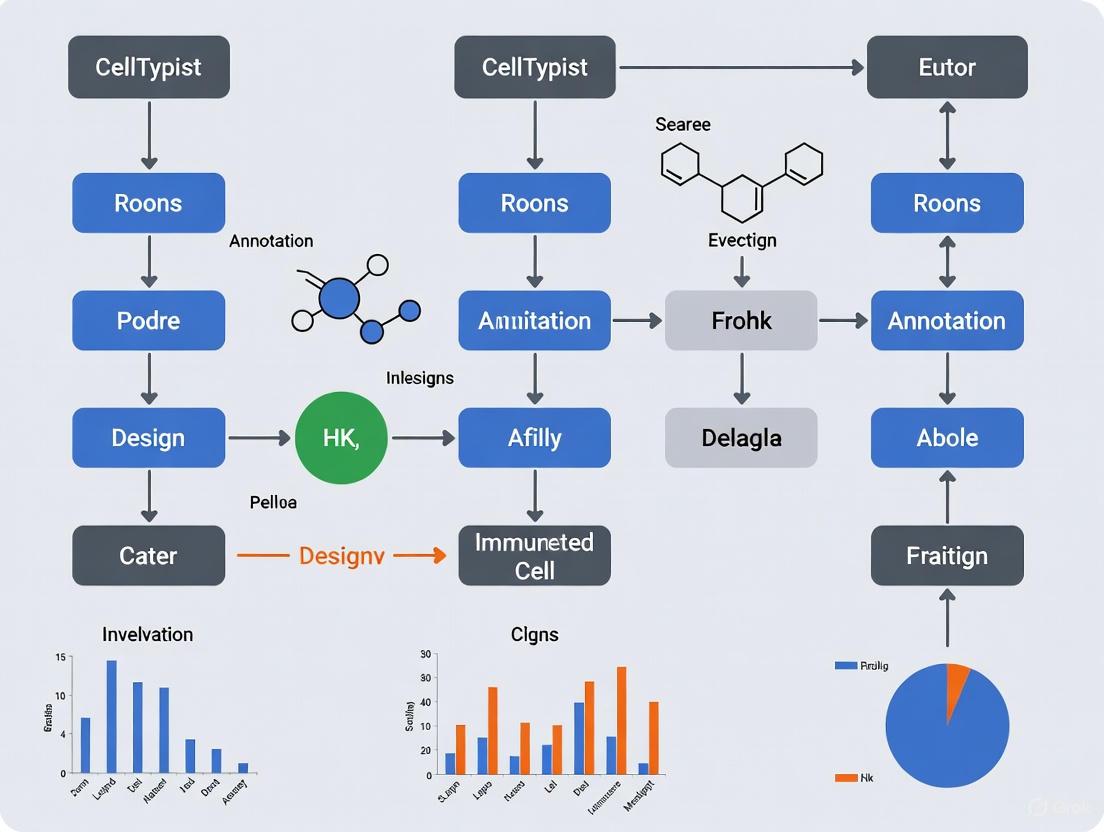
Workflow for Cell Type Annotation Using CellTypist
The following diagram illustrates the general workflow for applying CellTypist models to an scRNA-seq dataset, from data preparation to result interpretation.
Protocol 1: Model Inspection and Selection in Python
Before applying a model, it is good practice to inspect its contents. The following protocol details this process.
Step 1: Install and Import CellTypist Ensure CellTypist is installed in your Python environment. Then, import the necessary modules.
Step 2: Download the Models Download the models of interest to your local machine. The default storage directory is
~/.celltypist/.Step 3: Inspect Model Content Load a model and examine the cell types it contains to confirm it suits your research question.
Protocol 2: Cell Annotation and Result Refinement
This protocol covers the core annotation process and the optional but recommended majority voting refinement.
Step 1: Prepare Input Data Your input data should be a raw count matrix in a format like
.csvor.h5ad. For.h5adfiles, the data should be log-normalized (to 10,000 counts per cell) usingscanpy.pp.normalize_total(target_sum=1e4)andscanpy.pp.log1p()[8].Step 2: Run Cell Annotation Use the
celltypist.annotatefunction to predict cell identities. Themodeparameter allows you to choose between the "best match" (default) and a more conservative "probability match" strategy.Step 3: Apply Majority Voting Refinement Majority voting refines initial predictions by over-clustering the data and assigning the most frequent label within each local cluster to all its cells. This helps to reduce noise and improve consistency.
Step 4: Interpret and Export Results The prediction results can be examined, exported as tables, or converted into an
AnnDataobject for further analysis and visualization.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for scRNA-seq Annotation with CellTypist
| Item | Function / Description | Example / Note |
|---|---|---|
| CellTypist Python Package | Core software for automated cell type annotation. | Install via pip install celltypist [4]. |
Pre-trained Models (Immune_All_Low/High) |
Reference classifiers containing immune cell type signatures. | Downloaded via models.download_models() [1]. |
| Processed scRNA-seq Data | Query data in a compatible format for annotation. | A log-normalized count matrix in .h5ad or .csv format [8]. |
| Scanpy | Python library for single-cell data analysis. | Used for data pre-processing (normalization, PCA) and visualization (UMAP) [18]. |
| Jupyter Notebook / Python Script | Environment for executing the analysis workflow. | Provides reproducibility and a record of the analysis steps. |
Case Study: Application in a Multi-omic Immune Atlas
The utility of CellTypist's models is exemplified by their use in constructing the Human Immune Health Atlas, a high-resolution reference from over 100 healthy donors aged 11 to 65 years [18] [19]. In this large-scale study, researchers utilized multiple CellTypist models (Immune_All_High, Immune_All_Low, and Healthy_COVID19_PBMC) alongside Seurat's reference to guide expert annotation of 71 immune cell subsets [18]. This atlas, which includes 35 T cell, 11 B cell, 7 monocyte, and 6 NK cell subsets, was subsequently used to label cells in a longitudinal multi-omic study of immune aging [20] [19]. The project's analytical trace, from raw FASTQ files to the final annotated atlas, is documented within the Human Immune System Explorer (HISE) framework, showcasing a rigorous and reproducible application of these tools [18]. This real-world example demonstrates how CellTypist models can be integrated into a larger, high-throughput pipeline to generate biologically significant findings, such as identifying robust, non-linear transcriptional reprogramming in T cell subsets with age [20].
Troubleshooting and Best Practices
- Data Preprocessing is Critical: Ensure your query data is properly normalized. For the online interface,
.csvfiles require raw counts, while.h5adfiles require log-normalized data [8]. Mismatched normalization can lead to poor predictions. - Choosing Between Low and High Resolution: Begin your analysis with the
Immune_All_Lowmodel for the most detailed view. If the results appear overly fragmented or noisy for your research question, switch toImmune_All_Highto consolidate cells into broader, more robust populations. - Leverage Majority Voting: Always enable majority voting (
majority_voting = True) for your final analysis. This step is crucial for consolidating predictions within biologically meaningful clusters and mitigating the impact of outlier cells or technical artifacts [8]. - Biological Validation is Essential: Treat automated annotations as strong hypotheses. Use marker gene expression (e.g., via dot plots or feature plots in Scanpy) and prior biological knowledge to validate the assigned cell types, especially for rare or unexpected populations.
CellTypist's Role in the Single-Cell Analysis Workflow Pipeline
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological and medical research by enabling the exploration of transcriptomic profiles at individual cell resolution, revealing cellular heterogeneity and complex communication networks [21]. A critical step in scRNA-seq analysis is cell type annotation, which traditionally relied on manual expert knowledge, introducing subjectivity and variability [22]. CellTypist addresses these challenges as an automated cell type annotation tool specifically designed for scRNA-seq datasets, leveraging machine learning to provide rapid, precise classification of immune cell types and subtypes [4] [2].
This computational tool implements regularised linear models optimized via Stochastic Gradient Descent (SGD), balancing prediction accuracy with computational efficiency [4] [15]. Its training incorporates a comprehensive collection of immune cells from multiple tissues, creating a pan-tissue immune database that enables robust annotation across diverse biological contexts [2]. Unlike methods dependent on limited reference datasets, CellTypist's community-driven approach facilitates continuous knowledge expansion, allowing researchers to contribute new cell types and annotations [15].
Performance Characteristics and Quantitative Evaluation
Model Performance Metrics
CellTypist demonstrates high performance in cell type classification across multiple metrics. Validation studies reported precision, recall, and global F1-scores of approximately 0.9 for classification at both high- and low-hierarchy levels [2]. The tool's performance compares favorably against other label-transfer methods while maintaining minimal computational costs, making it suitable for large-scale datasets [2].
Table 1: Performance Metrics of CellTypist Models
| Metric | High-Hierarchy Model | Low-Hierarchy Model | Evaluation Context |
|---|---|---|---|
| Precision | ~0.9 | ~0.9 | Multi-tissue immune cell classification [2] |
| Recall | ~0.9 | ~0.9 | Multi-tissue immune cell classification [2] |
| F1-Score | ~0.9 | ~0.9 | Multi-tissue immune cell classification [2] |
| Training Cells | 360,000+ | 360,000+ | 16 tissues from 12 donors [2] |
| Cell Types Resolved | 32 | 91 | Initial model specifications [2] |
Comparison with Alternative Approaches
When benchmarked against emerging annotation methods, CellTypist maintains distinct advantages. A 2025 study evaluating LLM-based approaches found that while tools like LICT (Large Language Model-based Identifier for Cell Types) showed promise, CellTypist provided more consistent performance across diverse tissue contexts [22]. Specifically, LLM-based methods demonstrated diminished performance when annotating less heterogeneous datasets, with consistency rates dropping to 39.4% for embryo data and 33.3% for fibroblast data compared to manual annotations [22].
Table 2: Cross-Tool Performance Comparison in Immune Cell Annotation
| Tool | Methodology | Strengths | Limitations | Best Application Context |
|---|---|---|---|---|
| CellTypist | Logistic regression with SGD | High precision (~0.9), fast computation, immune-focused | Limited non-immune cell types | Multi-tissue immune cell annotation [4] [2] |
| LICT | Multi-model LLM integration | Reference-free, objective credibility assessment | Lower consistency in low-heterogeneity data (~39%) | Scenarios requiring reference-free approach [22] |
| Manual Annotation | Expert knowledge | Incorporates domain expertise | Subjective, time-consuming, variable between experts | Small datasets with available expertise [22] |
| GPTCelltype | Single LLM (ChatGPT) | No reference data needed | Limited biological context adaptation | Preliminary annotations before refinement [22] |
Experimental Protocols for CellTypist Implementation
Data Preparation and Input Requirements
Proper data preparation is essential for optimal CellTypist performance. The tool accepts multiple input formats, each with specific requirements:
- Format Specifications: Input data can be provided as
.csv,.h5ad,.txt,.tsv,.tab, or.mtxfiles [1]. For online analysis, only.csvand.h5adformats are accepted [8]. - Matrix Orientation: Expression matrices can be provided with cells as rows and genes as columns, or the reverse with appropriate parameter adjustment [1].
- Normalization Requirements: For
.csvfiles, raw count matrices are expected to reduce file size and upload burden. For.h5adfiles, log-normalized expression matrices (normalized to 10,000 counts per cell) are required, processed byscanpy.pp.normalize_total(target_sum=1e4)followed byscanpy.pp.log1p()[8]. - Gene Format: Gene symbols should be used as identifiers in the expression matrix [8].
Model Selection and Configuration
CellTypist provides multiple pre-trained models optimized for different annotation contexts:
- ImmuneAllLow.pkl: Recommended starting point for immune cell types, containing low-hierarchy (high-resolution) cell types and subtypes [14].
- ImmuneAllHigh.pkl: Alternative model with high-hierarchy (low-resolution) immune cell types [14].
- Custom Models: User-trained models for specialized applications [15].
Cell Annotation Workflow
The core annotation process involves transferring cell type labels from reference models to query data:
Result Interpretation and Validation
CellTypist generates multiple output matrices requiring different interpretation approaches:
- predicted_labels: Primary annotation results, including over-clustering information and majority-voting refined labels [8].
- decision_matrix: Continuous scores representing each cell's similarity to reference cell types [8].
- probability_matrix: Transformed probabilities (via sigmoid function) for each cell type assignment [8].
Integration with Single-Cell Analysis Workflows
Workflow Integration Diagram
The following diagram illustrates CellTypist's role within a comprehensive single-cell analysis pipeline:
Majority Voting Mechanism
CellTypist's majority voting refinement significantly improves annotation accuracy by leveraging transcriptional similarity among cells:
Table 3: Key Research Reagent Solutions for CellTypist Implementation
| Resource Category | Specific Solution/Format | Function in Workflow | Implementation Notes |
|---|---|---|---|
| Input Data Formats | .h5ad (AnnData) |
Preferred format for Python workflow | Contains log-normalized expression matrix [8] |
.csv (raw counts) |
Alternative for online interface | Required for web-based CellTypist analysis [4] | |
| Reference Models | Immune_All_Low.pkl |
High-resolution immune cell annotation | Recommended starting point [14] |
Immune_All_High.pkl |
Lower-resolution immune cell annotation | Alternative for broader classification [14] | |
| Software Dependencies | Python 3.6+ | Core programming environment | Required for local installation [4] |
| Scanpy | Single-cell analysis ecosystem | Enables seamless data exchange [1] | |
| NumPy/SciPy | Mathematical operations | Foundation for model calculations [15] | |
| Computational Resources | CPU configuration | Model application | Minimum 8GB RAM recommended for large datasets |
| Internet connection | Model download | Required for initial setup and updates [1] |
Advanced Applications and Integration Opportunities
Multi-Tissue Immune Cell Atlas Construction
CellTypist enables systematic resolution of immune cell heterogeneity across tissues, as demonstrated in a comprehensive analysis of 16 tissues from 12 donors [2]. This approach revealed tissue-specific features in mononuclear phagocytes, including distinct macrophage subpopulations in lung (alveolar macrophages expressing GPNMB and TREM2) and liver tissues [2]. The tool successfully classified 43 specific immune cell subtypes, including T cell subsets (CD4+ helpers, regulatory, cytotoxic), B cell compartments (naive, memory), and dendritic cell subsets (DC1, DC2, migDCs) [2].
Custom Model Development for Specialized Applications
For researchers investigating novel cell types or specialized tissues, CellTypist provides functionality for custom model development:
Integration with Spatial Transcriptomics
CellTypist annotations can be projected onto spatial transcriptomics data to resolve cellular organization patterns [21]. This integration enables mapping of immune cell distributions within tissue architecture, revealing spatial relationships between different immune subsets and their tissue microenvironments.
Troubleshooting and Technical Considerations
Common Implementation Challenges
- Gene Name Mismatches: Ensure consistent gene symbol nomenclature between query data and reference models [1].
- Batch Effects: While CellTypist demonstrates robustness to batch effects, pronounced technical variability may require integration methods prior to annotation [2].
- Novel Cell Types: The probability match mode (
mode = 'prob match') helps identify cells lacking clear reference counterparts [1]. - Computational Resources: Large datasets (>100,000 cells) may benefit from command-line implementation to optimize memory usage [4].
Quality Assessment Metrics
- Confidence Scores: Examine the
conf_scorefield in results to identify low-confidence predictions requiring manual verification [1]. - Marker Gene Expression: Validate annotations by checking expression of canonical marker genes for assigned cell types [2].
- Cross-Validation: Employ dataset splitting or cross-dataset validation to assess annotation stability [2].
CellTypist represents a robust, efficient solution for automated cell type annotation within single-cell RNA sequencing workflows, particularly for immune cell analysis. Its continuous model expansion and community-driven knowledge base position it as an increasingly valuable resource for the single-cell research community.
Cell Types, Subtypes, States, and Annotation Hierarchies
CellTypist is an automated cell type annotation tool for single-cell RNA sequencing (scRNA-seq) data that uses logistic regression classifiers optimized by a stochastic gradient descent algorithm [4] [1]. It represents a significant advancement in the field of cellular annotation by enabling rapid and consistent classification of immune cell types and subtypes without the subjectivity and time-intensive nature of manual annotation [23]. The platform operates through a global reference system that recapitulates cell type structure and biology across independent datasets, providing robust models that are both scalable and flexible for integration into existing analysis pipelines [4]. For researchers studying immune cells, CellTypist offers specially trained models with a current focus on immune sub-populations, allowing for accurate discrimination of closely related immune cell types [1].
The importance of automated annotation tools like CellTypist becomes evident when considering the limitations of manual annotation approaches, which can require 20 to 40 hours to manually annotate approximately 30 clusters in a typical single-cell dataset and are prone to subjective interpretation and inter-researcher variability [23]. Automated methods provide consistent results, enhance reproducibility, and significantly reduce analysis time while leveraging well-curated reference databases and computational algorithms [23]. CellTypist specifically addresses these challenges by implementing a supervised classification approach based on machine learning, where classifiers are trained using labeled reference scRNA-seq datasets and then applied to query datasets for cell type prediction [23].
Core Methodology of CellTypist
Algorithmic Foundation
CellTypist employs a regularized linear model with Stochastic Gradient Descent (SGD) to provide fast and accurate prediction of cell identities [4] [1]. The SGD optimization allows the model to efficiently handle large-scale scRNA-seq data while maintaining robust performance across diverse tissue types and experimental conditions. The model operates on raw count matrices (reads or UMIs) and requires gene expression data in either cell-by-gene or gene-by-cell format, supporting multiple file types including .txt, .csv, .tsv, .tab, .mtx or .mtx.gz [1]. A key aspect of the algorithmic implementation is the recommendation to include non-expressed genes in the input table as they provide important negative transcriptomic signatures that enhance the model's discriminatory power when compared against the reference model [1].
The prediction workflow in CellTypist offers two distinct modes for cell type assignment [1]:
- Best Match Mode (default): Each query cell is predicted into the cell type with the largest score/probability among all possible cell types. This approach works well for differentiating highly homogeneous cell types.
- Probability Match Mode: A more flexible approach where query cells are assigned to multiple cell types if they exceed a probability threshold (default: 0.5), or marked as "Unassigned" if they fail to pass the probability cutoff for any cell type. This mode accommodates cells with ambiguous identities or transitional states.
Model Architecture and Reference Databases
CellTypist employs a structured model architecture that includes both built-in and custom-trained models. The built-in models, such as Immune_All_Low.pkl and Immune_All_High.pkl, are specifically optimized for immune cell annotation and are regularly updated to incorporate the latest biological knowledge [1]. These models are distributed through a centralized repository, with each model file averaging approximately 1 megabyte in size, making them easily downloadable and manageable [1]. Users can access comprehensive information about available models through the models.models_description() function and download specific models or entire collections based on their research needs [1].
The model structure encapsulates detailed information about cell types and the features (genes) used for discrimination. Users can inspect any model by loading it as an instance of the Model class, which provides access to the complete set of cell types and genes/features contained within the model [1]. This transparency allows researchers to verify the biological relevance of the reference model before applying it to their data. By default, CellTypist stores these models in a folder called .celltypist/ within the user's home directory, though this location can be customized through environment variables [1].
Quantitative Performance Benchmarks
Comparison with Manual and Alternative Automated Methods
Recent benchmarking studies have demonstrated CellTypist's strong performance in automated cell type annotation. In comprehensive evaluations across diverse biological contexts, including normal physiology (PBMCs), developmental stages (human embryos), disease states (gastric cancer), and low-heterogeneity cellular environments (stromal cells), CellTypist and similar automated methods have shown consistent performance advantages over manual annotation approaches [22]. The tool's logistic regression framework combined with SGD optimization provides particularly robust performance for immune cell annotation, where it successfully discriminates between closely related cell subtypes [1].
Table 1: Performance Comparison of Cell Type Annotation Methods
| Method Category | Approach | Accuracy Range | Time Requirements | Consistency | Key Limitations |
|---|---|---|---|---|---|
| Manual Annotation | Expert-based marker inspection | Variable (subjective) | 20-40 hours for 30 clusters | Low inter-researcher consistency | Subjective, experience-dependent, time-consuming [23] |
| CellTypist | Logistic regression + SGD | High (immune cells) | Minutes to hours | High | Reference-dependent [1] |
| LLM-Based Methods (LICT) | Multi-model integration + talk-to-machine | 48.5-69.4% full match rate | Moderate | High | Performance varies by dataset heterogeneity [22] |
| sc-ImmuCC | Hierarchical + ssGSEA | 71-90% accuracy | Moderate | High | Specific to immune cells [24] |
Performance Across Dataset Types
CellTypist's performance demonstrates variability depending on the heterogeneity of the cell populations being analyzed. In highly heterogeneous datasets such as peripheral blood mononuclear cells (PBMCs) and gastric cancer samples, automated annotation tools typically achieve high accuracy with mismatch rates between 2.8% and 9.7% when compared to expert manual annotations [22]. However, in low-heterogeneity environments such as stromal cells in mouse organs or specific developmental stages in human embryos, the performance of all automated methods, including CellTypist, shows more variability, with match rates ranging from 43.8% to 48.5% for embryo and fibroblast data [22]. This pattern highlights the importance of dataset characteristics in determining the appropriate annotation approach and the potential need for method selection based on specific experimental contexts.
Table 2: Annotation Performance Across Biological Contexts
| Biological Context | Example Tissue/Cell Types | CellTypist Performance | Key Challenges | Recommended Approach |
|---|---|---|---|---|
| High Heterogeneity | PBMCs, Gastric Cancer | Mismatch rates: 7.5-9.7% [22] | Distinguishing closely related subtypes | Standard CellTypist models with best match mode |
| Low Heterogeneity | Embryonic cells, Stromal cells | Match rates: 43.8-48.5% [22] | Limited transcriptomic distinction | Ensemble methods + manual verification |
| Immune-specific | T cell subsets, B cell types | High accuracy for major subtypes [1] | Rare cell population detection | Specialized immune models + majority voting |
| Cross-tissue | Multiple organ systems | Recapitulates tissue-specific features [3] | Batch effects, technical variation | Batch correction + tissue-aware models |
Hierarchical Annotation Frameworks for Immune Cells
Conceptual Foundation of Annotation Hierarchies
Hierarchical annotation represents an advanced approach to cell type classification that mirrors the natural differentiation pathways of immune cells. This method organizes cell identities in a tree-like structure, with broad categories at higher levels (e.g., lymphoid vs. myeloid cells) and progressively finer subdivisions at lower levels (e.g., CD4+ T cell subsets) [24]. The hierarchical framework is particularly valuable for immune cells given their extensive diversity and lineage relationships, enabling more accurate and biologically meaningful annotations that capture both major cell types and specialized subtypes [24] [25]. Tools like sc-ImmuCC implement this approach through a three-layer hierarchy that can annotate nine major immune cell types and 29 cell subtypes, significantly improving annotation granularity compared to flat classification systems [24].
The power of hierarchical annotation lies in its ability to model the developmental continuum of immune cells while maintaining discrete classification categories that are practical for downstream analysis. This approach acknowledges that cells exist along differentiation trajectories rather than in strictly discrete categories, while still providing defined reference points for consistent annotation across datasets [26]. For T cells specifically, hierarchical frameworks have demonstrated the capacity to identify 46 reproducible gene expression programs (GEPs) reflecting core T cell functions including proliferation, cytotoxicity, exhaustion, and effector states, far exceeding the resolution of traditional clustering-based approaches [26].
Implementation in CellTypist
CellTypist implements hierarchical annotation through its majority voting feature, which refines initial cell-level predictions by considering the consensus of cells within clusters [4] [1]. This approach operates as a two-tiered hierarchy: first, individual cells receive preliminary annotations based on their transcriptomic profiles; second, these predictions are contextualized within cluster-level patterns to generate more robust assignments. The majority voting process significantly enhances annotation accuracy by leveraging the biological principle that cells of the same type tend to cluster together in transcriptional space, thereby reducing spurious assignments based on technical noise or individual cell variability [1].
The tool also supports custom model training, allowing researchers to build hierarchical classifiers tailored to specific biological questions or tissue types [1]. This flexibility enables the creation of specialized annotation frameworks that can capture tissue-specific immune cell states or disease-associated alterations in cell identity. For complex immune cell landscapes, such as tumor microenvironments, CellTypist's ability to integrate multiple reference models provides a pseudo-hierarchical approach that can resolve subtle differences between activated, exhausted, and resident memory T cell subsets [3].
Experimental Protocols for Immune Cell Annotation
Standard CellTypist Workflow
The standard CellTypist workflow for immune cell annotation involves sequential steps from data preparation through final annotation and visualization. The following protocol outlines the key experimental steps for comprehensive immune cell profiling:
Data Preprocessing Requirements:
- Input data must be a raw count matrix (reads or UMIs) in either cell-by-gene or gene-by-cell format
- Recommended file formats:
.csv,.h5ad,.txt,.tsv,.tab, or.mtx/.mtx.gz - For
.mtxformats, separate gene and cell files must be provided - Non-expressed genes should be included as they provide negative transcriptomic signatures [1]
Cell Type Prediction Protocol:
- Import CellTypist and available models:
- Download and inspect appropriate immune cell models:
- Perform cell type prediction with majority voting:
- Extract and examine results:
- Export results in multiple formats:
Validation and Quality Control:
- Compare annotations across different models (e.g., ImmuneAllLow vs. ImmuneAllHigh)
- Verify confidence scores for critical cell populations
- Inspect decision matrices for ambiguous assignments
- Visualize annotations in low-dimensional space (UMAP/t-SNE) to check for coherent labeling [1]
Advanced Hierarchical Annotation for T Cell Subsets
For researchers focusing specifically on T cell biology, advanced hierarchical approaches provide enhanced resolution of T cell states and functions. The following protocol integrates CellTypist with specialized T cell annotation frameworks:
T Cell-Specific Annotation Workflow:
- Primary T Cell Identification:
- Use CellTypist with immune models to isolate T cells from heterogeneous samples
- Apply probability match mode (p_thres = 0.5) to identify ambiguous cells
- Extract T cell populations for secondary analysis
Subset Resolution:
- Apply T cell-specific references or custom models
- Utilize hierarchical classification strategies
- Implement cross-dataset normalization when integrating multiple samples
Functional State Annotation:
- Integrate gene expression programs (GEPs) for activation, exhaustion, and differentiation states
- Employ tools like T-CellAnnoTator (TCAT) for predefined GEP quantification [26]
- Contextualize states within subset identities (e.g., exhausted CD8+ T cells vs. exhausted CD4+ T cells)
Validation and Biological Interpretation:
- Correlate transcriptomic annotations with surface protein expression (if available)
- Verify expected proportional relationships between subsets (e.g., CD4:CD8 ratios)
- Confirm presence of critical marker genes for assigned subsets
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Resources for Immune Cell Annotation
| Resource Category | Specific Tool/Resource | Function in Annotation Workflow | Key Features | Implementation in CellTypist |
|---|---|---|---|---|
| Reference Databases | ImmuneAllLow.pkl | Primary annotation of major immune cell types | Broad immune coverage, 1MB size [1] | Default model for immune annotation |
| Reference Databases | ImmuneAllHigh.pkl | High-resolution annotation of immune subtypes | Detailed subtype resolution [1] | Secondary model for validation |
| Custom Models | User-trained classifiers | Tissue-specific or disease-specific annotation | Tailored to specific research contexts [1] | celltypist.train() function |
| Analysis Environments | Python 3.6+ | Computational environment for CellTypist | Required for package installation [1] | Base requirement |
| Data Formats | .h5ad, .csv, .mtx | Input data compatibility | Flexible data input options [1] | Multiple format support |
| Visualization Tools | UMAP/t-SNE plotting | Visual validation of annotations | Spatial confirmation of labels [1] | predictions.to_plots() |
| Validation Resources | Decision matrices | Assessment of prediction confidence | Quantitative confidence scoring [1] | predictions.decision_matrix |
| Benchmarking Datasets | PBMC references | Method validation and performance testing | Standardized evaluation [22] | Quality control application |
Integration with Emerging Technologies
The field of automated cell type annotation is rapidly evolving with advancements in both sequencing technologies and computational methods. CellTypist exists within an ecosystem of complementary tools and approaches that collectively enhance our ability to resolve immune cell identities with increasing precision. Recent developments in large language model (LLM) applications for cell type annotation demonstrate promising alternative approaches that can achieve 48.5-69.4% full match rates with manual annotations across diverse datasets [22]. Tools like LICT (Large Language Model-based Identifier for Cell Types) leverage multi-model integration and "talk-to-machine" strategies to improve annotation reliability, particularly for challenging cell populations with ambiguous identities [22].
Single-cell long-read sequencing technologies represent another frontier with significant implications for cell type annotation, as they enable isoform-level transcriptomic profiling that provides higher resolution than conventional gene expression-based methods [27]. These technical advances offer opportunities to redefine cell types based on splicing patterns and isoform usage, potentially leading to more precise classifications of immune cell states and functions. Integration of these multi-modal data sources with tools like CellTypist will likely enhance annotation accuracy and biological relevance, particularly for discriminating between closely related immune cell states that exhibit subtle transcriptional differences.
For T cell immunology specifically, specialized annotation frameworks like TCAT (T-CellAnnoTator) and STCAT have demonstrated the ability to identify reproducible gene expression programs (GEPs) reflecting activation states, functional specializations, and subset identities [26] [25]. These tools can complement CellTypist by providing deeper insights into T cell biology beyond basic subset classification, enabling researchers to connect cell identities with functional capacities and clinical implications. The integration of these specialized approaches with CellTypist's robust classification framework represents a powerful strategy for comprehensive immune cell analysis in research and drug development contexts.
The Pan Immune Atlas represents a comprehensive, cross-tissue compendium of immune cells, systematically characterizing the diversity and distribution of immune populations across the human body. This atlas provides a foundational resource for understanding immune cell heterogeneity in health and disease, enabling the deconvolution of complex immune responses from various tissues [28]. Built upon large-scale single-cell RNA sequencing (scRNA-seq) initiatives, it captures detailed transcriptional profiles of both common and rare immune subsets, establishing a reference framework for automated cell type annotation.
CellTypist is an automated cell type annotation tool for scRNA-seq data that leverages logistic regression classifiers optimized by stochastic gradient descent (SGD) [11]. Its integration with immune cell atlases, including the Pan Immune Atlas, allows researchers to accurately classify immune cell types and subtypes in query datasets by leveraging pre-trained models built on comprehensive reference data [14] [4]. This synergy between expansive immune cell references and robust classification algorithms has positioned CellTypist as a valuable tool for standardized immune cell annotation in research and clinical applications.
Atlas Composition and Quantitative Features
The Pan Immune Atlas encompasses diverse immune cell populations across multiple biological contexts. The following table summarizes key quantitative features of major immune atlases integrated with CellTypist.
Table 1: Quantitative Features of Immune Cell Atlases in CellTypist
| Atlas/Model Name | Number of Cell Types/Subsets | Biological Context | Key Features | Source/Reference |
|---|---|---|---|---|
| Human Immune Health Atlas (Allen Institute) | 71 immune cell subsets [20] | Peripheral blood mononuclear cells (PBMCs) from healthy donors (age 25-90) [20] [29] | Cross-age atlas; >1.8 million cells from 108 healthy donors; longitudinal flu vaccination data [29] | Nature (2025) [20] |
| CellTypist Pan Immune Atlas v2 | 98 low-hierarchy cell types [17] | Multiple tissues; pan-immune system coverage [17] | Includes high- and low-hierarchy cell types; mapped to Cell Ontology IDs [17] | CellTypist Wiki [17] |
| Cross-Tissue Atlas | 76 non-epithelial cell subsets (majority immune) [28] | 35 healthy human tissues; 2.3 million cells [28] | Identified 12 cross-tissue coordinated cellular modules (CMs) [28] | Nature (2025) [28] |
The cellular composition of these atlases reveals significant immune heterogeneity across tissues. For example, the cross-tissue atlas demonstrated that peripheral blood and immune organs (bone marrow, lymph nodes, spleen) are predominantly composed of immune cells, while reproductive tissues exhibit a higher proportion of stromal cells [28]. Furthermore, rare subsets like age-associated B cells (ABCs), constituting less than 1% of total B cells, were identified not only in expected tissues like the liver and spleen but also in unexpected locations such as the ureter and skeletal muscle [28].
CellTypist Methodology for Immune Cell Annotation
Core Algorithm and Model Training
CellTypist operates on a logistic regression framework, with the option to implement SGD learning for large training datasets [14]. The model training process involves:
- Traditional Logistic Regression: Used in most cases for standard dataset sizes.
- SGD Logistic Regression: Applied for large datasets (>100,000 cells) using mini-batch training (1,000 cells per batch) over 10-30 epochs [14].
- Regularization: Incorporates L2 regularization to prevent overfitting and improve model generalizability.
- Gene Selection: Utilizes curated marker genes from the Pan Immune Atlas for feature selection in classification models [17].
Table 2: CellTypist Model Selection Guide for Immune Cell Annotation
| Model Name | Resolution | Cell Types Covered | Recommended Use Case |
|---|---|---|---|
| ImmuneAllLow | Low-hierarchy (High-resolution) | Detailed immune subtypes | Fine-grained annotation of immune cell subsets [14] |
| ImmuneAllHigh | High-hierarchy (Low-resolution) | Major immune lineages | Rapid annotation of major immune cell classes [14] |
| Pan Immune Atlas v2 | Multi-level | 98 immune cell types | Comprehensive cross-tissue immune annotation [17] |
Annotation Workflow and Validation
CellTypist Immune Annotation Workflow
The workflow begins with quality-controlled scRNA-seq data as input, followed by appropriate model selection based on the biological context of the query data [14]. For immune cell annotation, the "ImmuneAllLow" or "ImmuneAllHigh" models are typically recommended as starting points [14]. CellTypist then generates prediction probabilities for each cell, which can be further refined through majority voting to integrate predictions across similar cells and improve annotation robustness [4].
Experimental Protocols for Immune Cell Annotation
Basic Cell Annotation Protocol
Materials Required:
- Single-cell RNA sequencing data in matrix format (cells × genes)
- CellTypist installation (Python 3.6+ environment)
- Pre-trained immune cell model (downloaded automatically or manually)
Procedure:
Load data and import CellTypist in Python:
Download and select the appropriate immune model:
Run cell type prediction:
Examine and export results:
Advanced Protocol for Cross-Tissue Immune Annotation
For complex datasets involving multiple tissues or disease states, additional steps enhance annotation accuracy:
Model Customization:
- Combine multiple references to cover tissue-specific immune subsets
- Fine-tune models using transfer learning when reference data is limited
Hierarchical Annotation:
- First annotate major immune lineages using high-hierarchy models
- Then refine subsets using tissue-specific or state-specific models
Validation Integration:
- Incorporate marker gene expression validation post-annotation
- Compare with orthogonal methods (e.g., flow cytometry) when available
Table 3: Essential Research Reagents and Computational Tools for Immune Cell Annotation
| Resource Type | Specific Tool/Reagent | Function/Purpose | Availability |
|---|---|---|---|
| Reference Atlas | Human Immune Health Atlas [29] | Gold-standard reference for PBMC immune cells | Allen Institute Portal |
| Annotation Software | CellTypist [4] | Automated cell type classification | Python package: pip install celltypist |
| Cell Ontology | Cell Ontology IDs [17] | Standardized cell type nomenclature | Cell Ontology |
| Pre-trained Models | ImmuneAllLow, ImmuneAllHigh [14] | Ready-to-use classifiers for immune cells | Built-in CellTypist models |
| Validation Tool | LICT (LLM-based Identifier) [22] | Objective annotation reliability assessment | Communications Biology |
| Data Visualization | CellTypist UMAP Visualization [29] | Visual assessment of annotation quality | Allen Institute visualization tools |
Applications in Immunobiology and Drug Development
The integration of Pan Immune Atlas data with CellTypist annotation enables several advanced applications in research and therapeutic development:
Aging Immune Studies
Longitudinal immune profiling using CellTypist with the Human Immune Health Atlas has revealed non-linear transcriptional reprogramming in T cell subsets with age, particularly in naive CD4+ and CD8+ T cells, demonstrating robust changes prior to advanced aging [20]. These findings provide insights into age-related immune dysregulation that impacts vaccine responses and infection susceptibility.
Cancer Immunotherapy Research
CellTypist facilitates the analysis of tumor-infiltrating lymphocytes (TILs) using immune signatures derived from atlas data. Recent pan-cancer analyses have identified prognostic TIL signatures, such as the Zhang CD8 TCS signature, which demonstrates higher accuracy in prognostication across multiple cancer types [30]. This application enables better patient stratification for immunotherapy response.
Cross-Tissue Immune Coordination Analysis
The identification of coordinated cellular modules (CMs) across tissues reveals fundamental principles of immune organization. CellTypist can annotate these conserved cellular ecosystems, such as CM04 and CM05 enriched in primary and secondary immune organs, providing insights into systemic immune coordination and its dysregulation in disease [28].
Applications of CellTypist in Immune Research
Validation and Quality Control Framework
Ensuring annotation accuracy requires systematic validation approaches:
Objective Credibility Evaluation:
- Retrieve marker genes for predicted cell types
- Validate expression in corresponding clusters (>4 markers expressed in >80% of cells) [22]
- Calculate confidence scores based on marker concordance
Multi-Model Integration:
- Combine predictions from multiple LLMs or classification algorithms
- Leverage complementary strengths of different approaches
- Resolve discrepancies through iterative refinement [22]
Benchmarking Against Gold Standards:
- Compare with manual expert annotations
- Validate using orthogonal methods (e.g., protein markers)
- Assess reproducibility across technical replicates
This multi-layered validation framework addresses the limitations of both manual annotations (subjectivity, inter-rater variability) and automated methods (reference bias, technical artifacts), ensuring robust and biologically meaningful cell type assignments [22] [31].
Future Directions and Development
The continued expansion of Pan Immune Atlas resources and CellTypist capabilities includes several promising directions:
- Temporal Dynamics Integration: Incorporating longitudinal immune profiling data to model immune changes across lifespan and disease progression [20]
- Multi-omic Expansion: Integrating transcriptomic, proteomic, and epigenetic data for multi-modal cell type definition
- Spatial Context Integration: Combining single-cell resolution with spatial positioning information from emerging spatial transcriptomics technologies [28]
- Automated Ontology Alignment: Enhanced mapping to standardized cell ontologies to improve annotation consistency and interoperability [17]
These developments will further establish CellTypist as an essential tool for leveraging comprehensive immune cell atlases in basic research, translational studies, and therapeutic development.
Integration with Cell Ontology for standardized cell type identification
The advent of high-throughput single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to characterize cellular heterogeneity at unprecedented resolution. However, this technological advancement has introduced a significant challenge: the inconsistent annotation of cell types across different studies, tissues, and laboratories. The Cell Ontology (CL) serves as a structured, controlled vocabulary for cell types, providing standardized terminology and definitions that enable data integration and comparison across experiments. The integration of Cell Ontology with automated cell type annotation tools represents a critical advancement toward building a unified Human Cell Atlas where cellular annotations can be consistently interpreted across the scientific community [32].
CellTypist, an automated cell type annotation tool for scRNA-seq data, has emerged as a key platform that facilitates this integration. By incorporating Cell Ontology identifiers into its annotation framework, CellTypist enables researchers to bridge the gap between computational predictions and biologically meaningful, standardized cell type nomenclature. This integration is particularly valuable for immune cell annotation, given the extensive diversity and functional specialization of immune cell populations across tissues and physiological states [17]. The harmonization of cell type annotations through ontological frameworks addresses a fundamental challenge in single-cell biology - the reconciliation of annotation resolutions and technical biases across independently generated datasets [33].
Cell Ontology fundamentals and structure
The Cell Ontology is a community-based, structured vocabulary that represents a comprehensive collection of cell types across multiple species, with a particular emphasis on mammalian cell types. Built upon formal ontological principles, CL employs a directed acyclic graph structure where cell types are connected through "isa" and "partof" relationships, creating a hierarchical organization from broad to specific cell type categories. This hierarchical structure enables annotations at multiple levels of resolution, from general categories (e.g., "immune cell") to highly specialized subtypes (e.g., "CD4-positive, alpha-beta memory T cell") [32].
Each cell type in the CL is assigned a unique ontology identifier (e.g., CL:0000236 for B cells) and includes precise definitions, synonyms, and relationships to other cell types. This standardized approach facilitates computational reasoning and enables the integration of cell type information across different databases and analytical platforms. The CL is continuously updated through community curation efforts, incorporating new cell types as they are discovered and characterized through single-cell genomics and other experimental approaches [32].
For immune cells specifically, the CL encompasses the diverse lineages and functional states of the immune system, building upon decades of immunological research and classification systems such as the CD nomenclature established through the International Workshop on Human Leukocyte Differentiation Antigens [32]. This comprehensive coverage makes CL particularly suitable for standardizing annotations in immune cell-focused single-cell studies.
CellTypist's integration with Cell Ontology
Implementation of ontological standards
CellTypist incorporates Cell Ontology integration through several key mechanisms. The platform's model repository includes CL identifiers for the majority of cell types in its reference atlases, creating a direct mapping between computationally predicted labels and standardized ontological terms. This mapping enables consistent annotation across different datasets and analytical contexts [17]. For example, in the CellTypist Pan Immune Atlas v2, most low-hierarchy cell types are associated with specific CL identifiers, allowing predictions to be grounded in established biological definitions rather than dataset-specific nomenclature [17].
The integration occurs at both high-hierarchy (low-resolution) and low-hierarchy (high-resolution) levels, accommodating different analytical needs and biological questions. High-hierarchy categories represent broad cell classes (e.g., "T cells", "B cells"), while low-hierarchy categories capture more specialized subtypes (e.g., "Follicular B cells", "Memory B cells") [14]. This multi-level annotation approach aligns with the inherent hierarchical structure of the Cell Ontology, providing flexibility in annotation resolution while maintaining ontological standardization.
Validation of ontological mappings
A critical aspect of CellTypist's CL integration is the validation of ontological mappings. When CellTypist records are validated against the public Cell Ontology, the "Cell Ontology ID" column typically shows high validation rates, confirming that most identifiers correspond to valid terms in the current CL [17]. However, challenges can arise when comparing cell type names between CellTypist and the CL, as naming conventions may differ - for instance, CellTypist often uses plural forms ("B cells") while the CL typically uses singular forms ("B cell") [17].
Table 1: Cell Ontology Validation of CellTypist Annotations
| Validation Aspect | Performance | Common Issues | Resolution Approaches |
|---|---|---|---|
| Ontology ID Validation | High validation rate | Minimal issues with invalid IDs | Direct mapping to CL reference |
| Name-based Validation | Lower validation rate | Plural vs. singular conventions | Name standardization approaches |
| High-hierarchy Terms | 4 terms without ontology IDs | Terms like "B-cell lineage", "Cycling cells" | Community curation for missing terms |
| Synonym Recognition | 6 unique terms with detected synonyms | Alternative naming conventions | Synonym-aware mapping algorithms |
To address these discrepancies, CellTypist employs standardization approaches that include stripping trailing "s" characters from plural terms and leveraging synonym recognition, which significantly improves validation rates [17]. Additionally, the platform incorporates mechanisms to handle terms that lack direct CL counterparts, such as "B-cell lineage" and "Cycling cells," which remain without ontology identifiers and represent opportunities for future ontological expansion through community curation efforts [17].
Experimental protocols for ontology-integrated cell annotation
CellTypist installation and setup
To implement Cell Ontology-integrated cell type annotation using CellTypist, researchers must first establish the appropriate computational environment. CellTypist requires Python 3.6 or higher and can be installed via package managers such as pip or conda [4]:
Following installation, the essential Python packages must be imported, and the relevant CellTypist models should be downloaded:
The selection of appropriate models is crucial for obtaining biologically relevant annotations. For immune cell annotation, CellTypist provides specialized models such as "ImmuneAllLow" (for high-resolution annotation) and "ImmuneAllHigh" (for low-resolution annotation) [14]. These models incorporate Cell Ontology mappings, enabling standardized annotations across different levels of cellular resolution.
Data preparation and preprocessing
Proper data preparation is essential for robust cell type annotation. The query dataset should be formatted as an AnnData object, the standard data structure for single-cell data in the Python ecosystem. The data should undergo standard preprocessing steps including quality control, normalization, and highly variable gene selection:
It is critical to ensure that the gene identifiers in the query dataset match those in the CellTypist models. Typically, this involves using ENSEMBL gene IDs or standardized gene symbols to maximize compatibility and annotation accuracy [7].
Cell type annotation with ontological integration
The core annotation process involves applying CellTypist models to the preprocessed query data. The platform provides flexibility in annotation strategies, including the option to use majority voting to refine cell-level predictions into cluster-level annotations:
The resulting annotations include both cell type labels and associated Cell Ontology identifiers where available. Researchers can then map these annotations to the full Cell Ontology to access additional information such as formal definitions, relationships to other cell types, and marker genes [17].
Validation and interpretation of ontological annotations
Following annotation, researchers should validate the results through multiple approaches. First, the expression of canonical marker genes for the predicted cell types should be examined to confirm biological plausibility. Second, the distribution of Cell Ontology identifiers across the dataset should be analyzed to identify any terms that failed to validate or lack ontological mappings:
For cell types that lack proper ontological mappings or validate poorly, researchers may need to implement manual curation steps or contribute to community efforts to expand the Cell Ontology coverage for under-represented cell types [17].
Workflow visualization
Figure 1: Comprehensive workflow for Cell Ontology-integrated cell type annotation using CellTypist, showing the sequence from data preparation through validation and harmonization of annotations.
Table 2: Essential Research Reagents and Computational Resources for Cell Ontology-Integrated Annotation
| Resource Type | Specific Tool/Resource | Function in Workflow | Implementation Details |
|---|---|---|---|
| Software Tools | CellTypist Python Package | Automated cell type annotation | Install via pip/conda; requires Python 3.6+ [4] |
| Scanpy | scRNA-seq data preprocessing and analysis | Provides AnnData structure and analysis functions [7] | |
| Bionty | Cell Ontology validation and mapping | Enables ontology term validation and standardization [17] | |
| Reference Data | CellTypist Immune Models | Pre-trained models for immune cell annotation | ImmuneAllLow (120+ types) vs. ImmuneAllHigh (40+ types) [14] |
| Cell Ontology Database | Standardized cell type definitions and relationships | Provides hierarchical structure and relationship mappings [32] | |
| CellTypist Pan Immune Atlas | Comprehensive immune cell reference | Contains CL IDs for most cell types; community-curated [17] | |
| Input Data | scRNA-seq Count Matrix | Gene expression data for annotation | Format: .h5ad, .csv; requires appropriate gene identifiers [4] |
| Marker Gene Lists | Validation of annotation results | Canonical markers for immune cell types from literature [17] | |
| Validation Tools | CellHint | Cross-dataset harmonization | Resolves annotation differences across datasets [33] |
| popV | Ensemble annotation | Combines multiple classifiers with ontology-based voting [34] |
Validation and benchmarking of ontological annotations
Performance metrics and validation approaches
The integration of Cell Ontology with CellTypist annotations requires rigorous validation to ensure biological accuracy and computational robustness. Several approaches have been developed for this purpose, including marker-based validation where the expression of canonical marker genes is assessed for each annotated cell type, and cross-dataset validation where annotations are compared across multiple independent datasets [17] [34].
Benchmarking studies have demonstrated that CellTypist achieves high accuracy in immune cell annotation, particularly for well-characterized cell types such as classical monocytes, memory B cells, and CD8-positive alpha-beta memory T cells [34]. However, performance can vary for closely related cell types or transitional states, highlighting the importance of ontological standardization in resolving these ambiguous cases.
Table 3: Performance Metrics for CellTypist with Cell Ontology Integration
| Evaluation Dimension | Assessment Method | Typical Performance | Limitations and Considerations |
|---|---|---|---|
| Ontology ID Coverage | Percentage of annotations with valid CL IDs | High (>90% for immune cells) | Some high-hierarchy terms lack IDs [17] |
| Name Standardization | Validation of term names against CL | Moderate (improves with processing) | Plural/singular discrepancies require processing [17] |
| Cross-dataset Consistency | Harmonization across multiple datasets | Improved with CL integration | Technical batch effects can persist [33] |
| Resolution Appropriateness | Match between query data and model resolution | Model-dependent | High-resolution models require sufficient cell numbers [14] |
| Boundary Cell Handling | Annotation of cells between established types | Variable | Low consensus for transitional states [34] |
Comparison with alternative approaches
Several alternative tools exist for cell type annotation with varying approaches to ontological integration. CellHint employs predictive clustering trees to resolve cell-type differences in annotation resolution and technical biases across datasets, providing a relationship graph that hierarchically defines shared and unique cell subtypes [33]. popV implements an ensemble approach combining eight machine learning models with an ontology-based voting scheme to generate consensus annotations [34]. LICT leverages large language models in a "talk-to-machine" approach that iteratively refines annotations based on marker gene expression patterns [22].
Each approach has distinct strengths: CellTypist offers speed and interpretability through logistic regression models; CellHint specializes in cross-dataset harmonization; popV provides ensemble-based confidence scoring; and LICT enables reference-free annotation. The integration with Cell Ontology provides a common framework that facilitates comparison and integration of results across these different methodologies [33] [22] [34].
Advanced applications and future directions
Cross-tissue immune cell integration
The combination of CellTypist and Cell Ontology enables sophisticated analyses of immune cells across multiple tissues and physiological states. By providing a standardized framework for cell type identification, researchers can track specific immune populations across different anatomical sites, developmental stages, and disease conditions. This approach has revealed tissue-specific adaptations of immune cells while maintaining consistent classification through ontological standardization [33] [3].
For example, applications across 12 human tissues from 38 datasets have created a deeply curated cross-tissue database containing approximately 3.7 million cells with harmonized cell types, enabling the identification of rare immune populations and their distribution across the human body [33]. These resources provide unprecedented opportunities for understanding immune system organization and function at a whole-organism level.
Disease-specific annotation frameworks
In disease contexts, particularly cancer and autoimmune disorders, immune cell states can diverge significantly from healthy references. CellTypist's integration with Cell Ontology allows for the annotation of both canonical immune cell types and disease-associated states, facilitating the identification of clinically relevant immune populations [25]. For instance, in lung cancer samples, consistently enriched CD4+ Th17 cells have been identified in late-stage patients, while mucosal-associated invariant T (MAIT) cells were prevalent in milder cases of COVID-19 [25].
The ontological framework provides a structure for incorporating these disease-specific states while maintaining relationships to established cell types in the CL. This approach enables researchers to distinguish between fundamental cell lineages and contextual functional states, creating a more nuanced understanding of immune responses in disease.
Community expansion of ontological coverage
As single-cell technologies continue to reveal new cellular diversity, the Cell Ontology requires continuous expansion and refinement. CellTypist serves not only as a consumer of ontological standards but also as a platform for community-driven ontology development. Cell types identified through CellTypist that lack proper CL identifiers represent candidates for future ontological curation [17].
Researchers can contribute to this expansion by documenting novel cell types with sufficient evidence and submitting them to the Cell Ontology curation team. This collaborative cycle between computational tool development and ontological standardization represents a powerful paradigm for keeping pace with the rapid advances in single-cell biology.
CellTypist is an automated cell type annotation tool for single-cell RNA sequencing (scRNA-seq) data, leveraging logistic regression classifiers optimized by stochastic gradient descent (SGD) algorithm [4] [1]. Its machine learning framework enables rapid and precise cell type annotation, which is particularly valuable for dissecting immune cell heterogeneity across tissues [2]. This Application Note provides an updated overview of CellTypist's latest features and detailed protocols to empower researchers in immune cell annotation research.
Recent version updates and feature additions
Recent updates have significantly enhanced CellTypist's capabilities, with version 1.7.1 representing the latest stable release as of June 2025 [11]. The table below summarizes key developments in recent versions.
Table 1: Recent CellTypist version updates and features
| Version | Release Date | Key Updates and New Features |
|---|---|---|
| 1.7.1 | June 25, 2025 | Fixed command parameter error for GPU utilization [11]. |
| 1.7.0 | June 22, 2025 | Added GPU option for command line; introduced model subset function; adjusted Leiden parameters to parallel Scanpy [11]. |
| 1.6.3 | June 6, 2024 | Added GPU support based on rapids-singlecell for over-clustering; enabled direct loading of custom models [11]. |
| 1.6.1 | September 25, 2023 | Added cuML-based GPU support for model training; improved gene symbol to ID conversion [11]. |
A critical advancement in recent versions is the expanded GPU support, which accelerates computation-intensive steps like model training and over-clustering [11]. The implementation now supports both cuML-based training and rapids-singlecell for over-clustering, significantly speeding up analysis of large datasets.
Model ecosystem and selection strategy
CellTypist operates using a curated collection of pre-trained models, primarily focused on immune cell types [14] [15]. These models are trained using a logistic regression framework, with SGD optimization implemented for larger datasets using mini-batch training (1,000 cells per batch) [14] [15].
Table 2: Essential built-in models for immune cell annotation
| Model Name | Resolution | Cell Types | Recommended Use |
|---|---|---|---|
| ImmuneAllLow | Low hierarchy (High-resolution) | 91 cell types and subtypes | Fine-grained discrimination of immune cell subtypes [14] [2]. |
| ImmuneAllHigh | High hierarchy (Low-resolution) | 32 cell types | Initial exploration and major population annotation [14] [2]. |
The selection between high and low hierarchy models depends on the research question. High-resolution models (Immune_All_Low) are ideal for detecting rare cell populations and subtle phenotypic differences, while low-resolution models (Immune_All_High) provide a more robust overview of major cell lineages [14].
Computational workflow for cell annotation
The standard CellTypist workflow involves model downloading, data annotation, and result interpretation. The following diagram illustrates the core analytical pathway.
Figure 1: Core CellTypist analytical workflow for automated cell type annotation.
Model download and inspection
Before annotation, appropriate models must be downloaded and inspected. The following protocol details this critical first step.
Table 3: Protocol for model handling and inspection
| Step | Python Code | Purpose | Key Parameters |
|---|---|---|---|
| 1. List Models | models.models_description() |
Display available models with descriptions [1]. | - |
| 2. Download Model | models.download_models(model='Immune_All_Low.pkl') |
Download specific model for annotation [1]. | model: Model filename(s) to download. |
| 3. Load Model | model = models.Model.load(model='Immune_All_Low.pkl') |
Load model into memory for inspection [1]. | model: Name of downloaded model. |
| 4. Inspect Features | model.cell_typesmodel.features |
Examine cell types and genes in model [1]. | - |
Cell annotation and majority voting
Cell annotation is performed using the celltypist.annotate function. For enhanced accuracy, CellTypist implements a majority voting approach based on over-clustering cells using Leiden algorithm [15]. This process refines predictions by assigning the dominant cell type within each transcriptional subcluster.
Figure 2: Majority voting process which refines initial predictions through transcriptional clustering.
Essential research reagents and computational tools
Table 4: Key research reagent solutions for CellTypist workflow
| Reagent/Resource | Type | Function | Example/Format |
|---|---|---|---|
| Pre-trained Models | Computational | Reference signatures for cell prediction [14] | Immune_All_Low.pkl, Immune_All_High.pkl |
| Input Data | Experimental | Query scRNA-seq data for annotation [1] | .csv, .h5ad, .mtx (cell-by-gene or gene-by-cell) |
| CellTypist Package | Software | Core annotation algorithms and utilities [4] | Python 3.6+ package (PyPI or Bioconda) |
| GPU Resources | Hardware | Accelerate computation for large datasets [11] | NVIDIA GPUs with cuML/rapids-singlecell |
Advanced annotation modes
CellTypist provides two distinct prediction modes for different biological scenarios:
- Best match mode (
mode = 'best match'): Default setting assigning each cell to the single type with highest decision score [1]. - Probability match mode (
mode = 'prob match'): Assigns labels based on probability threshold (defaultp_thres = 0.5), allowing for "Unassigned" or multi-label classifications when cells exceed threshold for multiple types [1].
The probability match mode is particularly valuable for identifying novel cell types or handling transitional states that may express markers of multiple lineages.
CellTypist has evolved into a robust, feature-rich solution for automated cell annotation, with recent developments focusing on computational performance through GPU acceleration and enhanced analytical capabilities. Its regularly updated model ecosystem and standardized protocols enable researchers to efficiently extract meaningful biological insights from complex scRNA-seq datasets, particularly in the context of immune cell heterogeneity across tissues and conditions.
Hands-On CellTypist Workflow: From Installation to Advanced Analysis Modes
CellTypist is an automated cell type annotation tool designed specifically for single-cell RNA sequencing (scRNA-seq) data, leveraging logistic regression classifiers optimized by a stochastic gradient descent algorithm [1]. For researchers in immunology and drug development, this tool provides a critical resource for accurately identifying and classifying immune cell sub-populations within complex tissue samples. The platform functions as a scalable and flexible Python-based implementation that integrates seamlessly into existing single-cell analysis pipelines, enabling robust cell type prediction using either its built-in models (with a strong focus on immune cells) or custom models trained by users [4] [1]. This protocol outlines the comprehensive installation and setup procedures for CellTypist, ensuring researchers can quickly establish a functional environment for immune cell annotation research.
System Requirements and Pre-installation Checklist
Prerequisite Software and Specifications
Before installing CellTypist, ensure your system meets the following requirements. Adherence to these specifications guarantees compatibility and optimal performance.
- Python: Version 3.6 or higher is required. Verify your Python version by running
python --versionin your terminal. - Package Manager: Either
pip(Python's package installer) orconda(package manager from the Anaconda/Miniconda distribution) must be available on your system. - Operating System: CellTypist is cross-platform and supports Windows, macOS, and Linux environments.
- Internet Connection: Required for the initial installation and for downloading model files.
- Disk Space: Reserve a minimum of 2-3 GB of free disk space for the software and its associated model files.
Research Reagent Solutions
The table below details the key computational "reagents" required for using CellTypist effectively in an immune cell annotation workflow.
Table 1: Essential Research Reagent Solutions for CellTypist
| Item Name | Function/Description | Usage in Workflow |
|---|---|---|
| CellTypist Python Package | Core software for automated cell type annotation. | Provides the primary logistic regression classifier for predicting cell types from gene expression data. |
Built-in Reference Models (e.g., Immune_All_Low.pkl) |
Pre-trained classifiers on curated immune cell datasets. | Serves as a reference for annotating query datasets. "Low" and "High" indicate resolution hierarchy [14]. |
| Scanpy | Python toolkit for single-cell data analysis. | Used for general data manipulation, normalization, and visualization integrated with CellTypist outputs [7]. |
| scRNA-seq Dataset | Input data for annotation, in formats like .csv, .h5ad, or .mtx. |
The query dataset containing raw UMI counts from immune cells to be classified. |
Environment File (environment.yml) |
Conda environment configuration file. | Ensures reproducible installation of CellTypist and all its dependencies with correct versions. |
Installation Procedures
This section provides a detailed, step-by-step protocol for installing CellTypist using two different package managers, giving researchers flexibility based on their existing setup.
Installation viapip
The pip installation method is straightforward and recommended for users who already have a Python environment configured.
- Open a terminal (Command Prompt on Windows, Terminal on macOS/Linux).
- Execute the installation command:
- Wait for the process to complete.
pipwill automatically fetch the CellTypist package from the Python Package Index (PyPI) and install it along with all necessary dependencies, such aspandas,scikit-learn,scanpy, andnumpy[4] [7].
Installation viaconda
The conda method is ideal for managing complex software environments and dependencies, reducing potential conflicts.
- Open a terminal.
- Execute the installation command from the
biocondachannel: The-c bioconda -c conda-forgeflags specify the channels from which the package and its dependencies should be retrieved [4] [35]. - Wait for conda to solve the environment and proceed with the installation as prompted.
Quantitative Comparison of Installation Methods
The table below summarizes the key characteristics of both installation methods to guide your choice.
Table 2: Comparison of CellTypist Installation Methods
| Feature | pip Installation | conda Installation |
|---|---|---|
| Command | pip install celltypist |
conda install -c bioconda -c conda-forge celltypist |
| Dependency Management | Uses PyPI; may require manual conflict resolution. | Uses conda ecosystems; superior for handling complex dependencies. |
| Environment Isolation | Relies on external tools like venv. |
Native environment isolation and management. |
| Recommended Use Case | Standard Python installations and virtual environments. | Anaconda/Miniconda users and complex project environments. |
| Source | [4] [11] | [4] [35] |
Environment Configuration and Model Setup
After successful installation, the next critical step is to configure the environment and download the necessary reference models for cell type annotation.
Downloading Reference Models for Immune Cell Annotation
CellTypist relies on pre-trained models for prediction. These models are downloaded on-the-fly and stored locally. The following Python code demonstrates how to download models.
By default, models are stored in a folder named .celltypist/ within the user's home directory. You can customize this location by setting the CELLTYPIST_FOLDER environment variable in your shell configuration file (e.g., ~/.bash_profile or ~/.bashrc):
Validating the Installation and Model
To confirm that CellTypist and its models are installed correctly, run a verification script.
A successful validation will display a summary of the model, a list of cell types (e.g., CD4+ T cells, CD8+ T cells, B cells, monocytes), and the gene list used for classification.
Workflow Diagram: Installation and Setup
The diagram below visualizes the complete installation and initial setup workflow for CellTypist, providing a logical map from setup to first use.
Basic Usage Protocol for Immune Cell Annotation
Following the installation and setup, this basic protocol demonstrates a standard CellTypist workflow for annotating a single-cell dataset containing immune cells.
- Data Preparation: Prepare your input data as a count table (cells-by-genes or genes-by-cells) in a supported format (e.g.,
.csv,.h5ad). A raw count matrix is required [1]. Cell Type Prediction: Use the
celltypist.annotatefunction to predict cell identities. Themajority_votingoption can be enabled to refine predictions based on cell clusters [4].Result Examination: The results are stored in an
AnnotationResultobject. Key outputs can be inspected and exported.
This workflow reliably assigns immune cell identities, forming a foundation for downstream biological interpretation and discovery in immunology research.
Within the framework of immune cell annotation research, the accurate preparation of single-cell RNA sequencing (scRNA-seq) data is a critical prerequisite for achieving biologically meaningful results with CellTypist. This protocol details the specific input formats and preprocessing requirements necessary to optimize the performance of CellTypist for the identification and characterization of immune cell subsets. Proper data preparation ensures that the logistic regression classifiers within CellTypist can effectively recapitulate the immune cell type structure and biology of independent datasets [4].
Input Data Formats
CellTypist accepts scRNA-seq data in multiple common formats, providing flexibility for researchers. The choice of format often depends on the size of the dataset and the analysis workflow.
Table 1: Supported Input File Formats and Their Specifications
| Format | Data Structure | Preprocessing State | Additional Files Required | Key Considerations |
|---|---|---|---|---|
| CSV (.csv) | Cell-by-gene (preferred) or gene-by-cell [8] [36] | Raw count matrix [8] | None | Ideal for smaller datasets; gene symbols as columns are required [36]. |
| H5AD (.h5ad) | AnnData object [36] | Log-normalized expression matrix [8] | None | Efficient for larger datasets; requires prior normalization [8]. |
| MTX (.mtx) | Matrix Market format [1] [36] | Raw count matrix [1] | Gene and cell files [1] [36] | Used for sparse matrix representation; requires transpose_input=True for gene-by-cell [36]. |
| Text Files | Cell-by-gene or gene-by-cell [1] | Raw count matrix [1] | None | Includes .txt, .tsv, .tab; similar considerations as CSV [1]. |
The fundamental requirement across all formats is a count matrix representing gene expression. CellTypist expects genes to be represented by gene symbols [36]. It is also recommended to include non-expressed genes in the input table, as they provide information on negative transcriptomic signatures valuable for the classification model [1].
Preprocessing Requirements
Data preprocessing is a critical step to ensure the input data is compatible with CellTypist's models, which are trained on data processed in a specific manner. The requirements differ slightly between the two primary file formats.
Preprocessing for CSV Input
For CSV files, a raw count matrix (either UMI counts or reads) is expected as input [1] [8]. This recommendation is made to reduce file size and the burden of online uploads. While CellTypist is robust to various data transformations, providing raw counts allows the tool to apply its own standardized processing pipeline, ensuring consistency with the model's training data [4].
Preprocessing for H5AD Input
In contrast, when using the H5AD format, the AnnData object should contain a log-normalized expression matrix [8]. This normalization should target a total count of 10,000 per cell, followed by a log1p transformation, as implemented by scanpy.pp.normalize_total(target_sum=1e4) and scanpy.pp.log1p() [8] [37]. The following code block demonstrates a typical preprocessing workflow using Scanpy for data destined for CellTypist in H5AD format.
Workflow Diagram
The following diagram summarizes the data preparation and preprocessing workflow for CellTypist.
Experimental Protocol for Immune Cell Annotation
This section provides a detailed, step-by-step protocol for annotating immune cells in a PBMC dataset using CellTypist, based on a demonstrated methodology [37].
Research Reagent Solutions
Table 2: Essential Materials and Reagents
| Item Name | Function / Description | Example / Source |
|---|---|---|
| CellTypist Python Package | Automated cell type annotation tool. | Install via pip install celltypist [4]. |
| Scanpy Package | Single-cell data analysis in Python. | Used for data loading, normalization, and preprocessing [37]. |
| Reference Model | Pre-trained classifier for immune cell types. | Immune_All_Low.pkl (high-resolution) or AIFI_L2.pkl (atlas-specific) [14] [37]. |
| scRNA-seq Dataset | Input data for annotation. | A cell-by-gene raw count matrix in a supported format (e.g., CSV, H5AD) [8]. |
Step-by-Step Procedure
Environment Setup and Package Installation
Load and Preprocess the Input Data
- If starting from a raw count matrix (e.g., 10X H5 file):
- Note: If your data is already preprocessed and saved as an H5AD file, you can load it directly with
sc.read_h5ad().
Download and Select a Reference Model
- CellTypist provides multiple built-in models. For comprehensive immune cell annotation, start with the general immune model or select a tissue-specific one.
Run CellTypist for Cell Annotation
Integrate Predictions and Visualize Results
- Transfer the prediction results into the AnnData object for downstream analysis and visualization.
Result Interpretation
Accessing and selecting appropriate models for immune cell annotation
CellTypist represents a transformative approach to automated cell type annotation for single-cell RNA sequencing (scRNA-seq) datasets, employing logistic regression classifiers optimized through stochastic gradient descent algorithms [15]. This tool has become particularly valuable for immune cell annotation, given the extensive diversity and complex heterogeneity of immune cell populations across tissues and disease states. As scRNA-seq technologies continue to advance, generating increasingly large datasets, the need for robust, scalable, and accurate annotation methods has become paramount in immunological research [27]. CellTypist addresses this need through regularly updated model repositories, flexible training options, and optimized computational frameworks that balance accuracy with efficiency [14] [11].
The importance of CellTypist in immune cell annotation research stems from its ability to recapitulate cell type structure and biology across independent datasets, providing a global reference framework that maintains consistency while accommodating tissue-specific variations [4]. For researchers and drug development professionals, this tool offers a standardized approach to cell type identification that can be integrated into existing analytical pipelines, facilitating comparative analyses and meta-analyses across studies and experimental conditions [1].
Accessing CellTypist models
Installation and basic setup
CellTypist can be installed through two primary package management systems, making it accessible across different computational environments [1] [11]:
Following installation, users can import CellTypist and access its model repository within a Python environment [1]:
Model download procedures
CellTypist provides multiple approaches for accessing its collection of pre-trained models, which are particularly valuable for immune cell annotation [1]:
Listing available models: The
models.models_description()function displays all available models that can be downloaded and used, providing researchers with an overview of their options before selection.Targeted model download: Specific models can be downloaded individually or as a group based on research needs:
Comprehensive model download: Since each model averages only 1 megabyte (MB) in size, users can download all available models to ensure full access to CellTypist's annotation capabilities:
Model updates: To ensure access to the most current models, users can force updates through the
force_update = Trueparameter, which re-downloads the latest versions [1].
By default, models are stored in a folder called .celltypist/ within the user's home directory. Advanced users can customize this storage location by setting the CELLTYPIST_FOLDER environment variable in their shell configuration file [1].
Model inspection and evaluation
Before applying models to query data, researchers can inspect model contents to ensure appropriate selection [1]:
This inspection capability allows researchers to verify that their genes of interest are represented in the model and that the cell types included align with their research questions.
Model selection framework for immune cell annotation
Hierarchy-based model selection
For immune cell annotation, CellTypist offers specialized models organized according to cellular resolution [14]:
Table 1: Hierarchy-based immune cell annotation models in CellTypist
| Model Name | Resolution | Hierarchy Level | Use Case | Number of Cell Types |
|---|---|---|---|---|
Immune_All_Low.pkl |
Low | High-resolution | Fine-grained immune cell subtype discrimination | To be determined from model inspection |
Immune_All_High.pkl |
High | Low-resolution | Broad immune cell categorization | To be determined from model inspection |
The "Low" hierarchy models provide high-resolution classification, enabling discrimination of closely related immune cell subtypes, while "High" hierarchy models offer broader categorization suitable for initial dataset exploration or when working with datasets where fine-grained distinctions may not be necessary [14]. This hierarchical approach allows researchers to match the annotation resolution to their specific research objectives and data quality.
Contextual model selection criteria
Beyond hierarchical level, researchers should consider several additional factors when selecting appropriate models for immune cell annotation:
Tissue context: While immune cells can circulate throughout the body, tissue-specific adaptations may influence gene expression patterns. Researchers should consider whether their data derives from specific tissues or systemic sources.
Species compatibility: Although not explicitly detailed in the available resources, researchers should verify model compatibility with their experimental species when applying CellTypist to non-human data.
Technical considerations: Model performance may vary based on sequencing platform, library preparation methods, and data quality. Researchers working with novel protocols should validate annotations through independent methods.
Biological context: Disease states, developmental stages, and experimental manipulations can alter cellular identities and expression profiles. Researchers should consider whether available models adequately capture these contextual variations.
Figure 1: Decision framework for selecting appropriate immune cell annotation models in CellTypist
Comprehensive annotation protocol
Data preparation and input formats
CellTypist accepts multiple input formats for query data, providing flexibility for researchers working with diverse data structures [1]:
Count table formats: CellTypist supports
.txt,.csv,.tsv,.tab,.mtx, or.mtx.gzfiles containing raw count matrices (reads or UMIs). A cell-by-gene format is preferred, though gene-by-cell matrices can be accommodated with thetranspose_input = Trueparameter.Data requirements: The input should represent raw count data without normalization. Importantly, non-expressed genes should be included in the input table as they provide negative transcriptomic signatures valuable for discrimination between cell types [1].
Matrix file considerations: When using
.mtxformat, researchers must additionally specifygene_fileandcell_filearguments to provide names of genes and cells, respectively.
Basic cell type annotation
The core annotation function in CellTypist employs the following protocol [1]:
If the model argument is not specified, CellTypist defaults to the Immune_All_Low.pkl model, reflecting its utility as a starting point for immune cell annotation [1].
Annotation modes
CellTypist offers two distinct annotation modes to address different research scenarios:
Best match mode (default): Each query cell is predicted to belong to the cell type with the largest score/probability among all possible cell types. This approach works well for distinguishing highly homogeneous cell types [1].
Probability match mode: This approach applies a probability cutoff (default: 0.5) to determine cell type assignments, allowing for unassigned cells or multi-label classification:
In this mode, query cells receive the label "Unassigned" if they fail to pass the probability cutoff for any cell type, or multiple labels concatenated by "|" if more than one cell type passes the probability threshold [1]. This capability is particularly valuable for identifying potentially hybrid cell states or poorly characterized immune populations.
Majority voting refinement
CellTypist incorporates a majority voting approach that refines initial predictions by leveraging the transcriptional similarity of cells within clusters [15]. This process operates on the principle that transcriptionally similar cells are more likely to form coherent subclusters regardless of their individual prediction outcomes.
The majority voting process involves the following steps [4]:
Over-clustering: The query data is over-clustered using Leiden clustering with a canonical Scanpy pipeline.
Consensus identification: Each resulting subcluster is assigned the identity supported by the dominant cell type predicted among its constituent cells.
Label refinement: Through this process, distinguishable small subclusters receive distinct labels, while homogeneous subclusters converge to consistent labels within larger clusters.
This approach can be activated with a simple parameter addition [4]:
Result interpretation and advanced analyses
Output examination and visualization
CellTypist returns an AnnotationResult object containing comprehensive prediction information [1]:
For visualization and further analysis, results can be transformed into an AnnData object with predicted labels and confidence scores embedded into observation metadata [1]:
CellTypist also provides direct visualization capabilities without explicit transformation to AnnData [1]:
Confidence assessment and validation
To evaluate prediction reliability, researchers can examine confidence scores and probability distributions:
This generates additional figures displaying decision scores and probability distributions for each cell type across the UMAP, helping researchers identify regions of high confidence or ambiguous assignments that may require further investigation [1].
Table 2: Key output components of CellTypist annotation
| Output Component | Description | Interpretation | Utility |
|---|---|---|---|
predicted_labels |
Final cell type assignments for each cell | Best match or probability-based assignment | Primary result for downstream analysis |
decision_matrix |
Raw decision scores for each cell-cell type pair | Linear combination of scaled expression and model coefficients | Technical assessment of model performance |
probability_matrix |
Probabilities transformed via sigmoid function | Normalized probabilities between 0-1 | Interpretable confidence metrics |
conf_score |
Confidence scores embedded in AnnData | Probability-based confidence for final assignment | Quality control and filtering |
Custom model training for specialized applications
Training protocol for immune cell models
When built-in models do not adequately address specific research needs, CellTypist enables training of custom models through the celltypist.train function [13]. This capability is particularly valuable for researchers working with specialized immune cell populations, novel immunological contexts, or unique experimental conditions.
The basic training protocol requires an input count matrix (X) and corresponding cell type labels [13]:
The input matrix can be provided as a path to a count table file, an AnnData object, or any array-like object already loaded in memory. When using an AnnData object as input, the labels argument can be specified as a column name from the cell metadata [13].
Optimization approaches for training
CellTypist offers multiple optimization strategies to accommodate datasets of different sizes and characteristics [13]:
Traditional logistic regression: Suitable for small to medium-sized datasets, with customizable regularization strength (
C) and solver options.Stochastic Gradient Descent (SGD) learning: Recommended for large datasets (>100k cells), with optional mini-batch training for improved efficiency.
Feature selection: Optional two-pass training with initial feature selection based on regression coefficients, which can improve model performance and reduce noise.
Table 3: Key parameters for custom model training in CellTypist
| Parameter | Default Value | Description | Impact on Model Performance |
|---|---|---|---|
use_SGD |
False |
Whether to implement SGD learning | Essential for large datasets; improves scalability |
mini_batch |
False |
Whether to implement mini-batch training | Further enhances efficiency for very large datasets |
balance_cell_type |
False |
Whether to balance cell type frequencies | Can improve recognition of rare cell populations |
feature_selection |
False |
Whether to perform feature selection | May enhance model generalizability and reduce overfitting |
top_genes |
300 |
Number of top genes selected per cell type | Balances feature richness against potential noise |
alpha |
0.0001 |
L2 regularization strength for SGD | Larger values may improve generalization at cost of accuracy |
C |
1.0 |
Inverse of L2 regularization for traditional LR | Smaller values may improve generalization |
Training workflow for immune cell models
Figure 2: Custom model training workflow in CellTypist for immune cell annotation
The scientist's toolkit for CellTypist implementation
Table 4: Essential research reagents and computational resources for CellTypist implementation
| Resource Category | Specific Tool/Resource | Function in Workflow | Implementation Notes |
|---|---|---|---|
| Computational Environment | Python 3.6+ | Base programming environment | Required for CellTypist installation and execution |
| Scanpy ≥1.7.0 | Single-cell analysis ecosystem | Enables seamless integration with CellTypist | |
| scikit-learn ≥0.24.1 | Machine learning backend | Powers the logistic regression algorithms | |
| Data Input Formats | Raw count matrices (cell-by-gene) | Primary input format | Required for model training and annotation |
| Cell type labels | Training data requirement | Essential for custom model development | |
| Pre-trained Models | Immune_All_Low.pkl |
High-resolution immune annotation | Default model for immune cell typing |
Immune_All_High.pkl |
Low-resolution immune annotation | Alternative for broad categorization | |
| Validation Tools | UMAP visualization | Result verification | Enables spatial assessment of predictions |
| Marker gene expression | Biological validation | Confirms annotation biological plausibility |
CellTypist represents a robust, scalable solution for immune cell annotation in scRNA-seq studies, offering both pre-trained models specifically optimized for immune populations and flexible custom training capabilities. Through its logistic regression framework optimized via stochastic gradient descent, CellTypist balances computational efficiency with biological accuracy, making it suitable for datasets ranging from focused immunological studies to large-scale atlas projects [14] [15].
The model selection framework presented in this protocol emphasizes the importance of matching annotation resolution to research objectives, with the Immune_All_Low.pkl and Immune_All_High.pkl models providing complementary approaches for high-resolution subtyping and broad categorization, respectively [14]. The integration of majority voting further enhances annotation accuracy by leveraging transcriptional similarity within clusters, while probability-based assignment modes accommodate ambiguous cases or potential novel cell states [1] [4].
For advanced applications, CellTypist's custom training capabilities enable researchers to develop specialized models tailored to unique immunological questions, experimental conditions, or novel cell populations [13]. This flexibility ensures that CellTypist can evolve alongside the rapidly advancing field of immunology and single-cell technologies.
As single-cell technologies continue to generate increasingly complex and multidimensional datasets, tools like CellTypist will play an essential role in extracting biologically meaningful insights through standardized, reproducible, and validated cell type annotation protocols.
CellTypist's basic prediction mode employs a best-match classification approach designed for automated cell type annotation of single-cell RNA sequencing (scRNA-seq) data. This mode utilizes regularized logistic regression models trained on extensively curated reference datasets to predict cell type identities based solely on gene expression patterns [4]. The fundamental principle underlying this approach is that cells of the same type will exhibit similar transcriptional profiles, enabling the model to assign the most probable cell type label to each query cell through a supervised classification framework [38]. This method is particularly effective for annotating homogeneous cell populations where distinct transcriptional signatures exist between cell types, making it invaluable for initial dataset characterization and immune cell annotation workflows [39].
Unlike more complex neural network architectures or integration-based methods, CellTypist's basic prediction mode implements a linear classification framework that provides transparent and interpretable results [4]. In scenarios involving homogeneous cell types, this approach has demonstrated performance comparable to nonlinear models while maintaining computational efficiency and lower resource requirements [38]. The basic prediction mode serves as the foundation for CellTypist's automated annotation pipeline, offering researchers a robust starting point for cell type identification before applying more sophisticated refinement techniques such as majority voting [8].
Model Specifications and Performance Metrics
Immune Cell Annotation Models
CellTypist provides specialized models pre-trained on immune cells collected from diverse tissues [14]. The selection between high-resolution and low-resolution models depends on the research question and the expected heterogeneity of the sample.
Table 1: CellTypist Immune Cell Models for Basic Prediction Mode
| Model Name | Resolution | Cell Types | Training Cells | Best For |
|---|---|---|---|---|
| ImmuneAllLow | Low (High-resolution) | 85 subtypes | ~200K | Detailed immune profiling, heterogeneous samples |
| ImmuneAllHigh | High (Low-resolution) | 15 major types | ~200K | Broad classification, initial screening |
The ImmuneAllLow model provides finer granularity, distinguishing between closely related immune cell subtypes such as CD4+ naive T cells, CD4+ central memory T cells, and CD4+ effector memory T cells [14]. This model is particularly valuable when investigating specific immune responses or characterizing diverse immunological niches. Conversely, the ImmuneAllHigh model offers broader categorization into major immune lineages (T cells, B cells, NK cells, monocytes, etc.), making it suitable for initial dataset exploration or when working with samples containing well-established immune populations [14].
Performance Benchmarks
In controlled benchmarking studies, CellTypist's logistic regression-based basic prediction mode has demonstrated robust performance across diverse immune cell types [38]. The model's accuracy scales with both training dataset size and model complexity, with the current implementation optimized for the reference corpus size.
Table 2: Performance Metrics for Basic Prediction Mode
| Metric | Performance | Notes |
|---|---|---|
| Overall accuracy | >85% | Varies by cell type specificity |
| Rare cell detection | Moderate | Depends on training representation |
| Computational speed | Fast | ~2000 cells/minute on standard CPU |
| Memory usage | Efficient | <8GB for 100K cells |
Performance is generally highest for well-defined immune populations with distinct transcriptional signatures, such as T cells versus B cells, while more closely related subsets (e.g., naive versus memory T cells) may show lower discrimination accuracy [39]. The model's decision threshold can be adjusted based on the required confidence level for downstream applications, with higher thresholds providing more conservative predictions at the potential cost of leaving more cells unclassified [7].
Experimental Protocol for Basic Prediction Mode
Data Preprocessing Requirements
Proper data preprocessing is critical for optimal performance of CellTypist's basic prediction mode. The protocol requires a log-normalized expression matrix with consistent gene symbol annotation [8].
Input Preparation Steps:
- Normalization: Normalize raw counts to 10,000 counts per cell using
scanpy.pp.normalize_total(target_sum=1e4)followed by logarithmic transformation withscanpy.pp.log1p()[8] - Gene Format: Ensure genes are specified using official HGNC symbols in the matrix
- Matrix Orientation: Format with cells as rows and genes as columns for .csv files, or standard AnnData orientation for .h5ad files
- Quality Control: Remove low-quality cells and doublets prior to analysis using standard scRNA-seq QC metrics
For the basic prediction mode, feature selection is handled internally by the model, which uses the same genes employed during model training. This eliminates the need for user-driven variable gene selection, streamlining the annotation workflow [4].
CellTypist Execution Protocol
The basic prediction mode can be executed through either Python API or command-line interface, with the following standardized protocol:
The basic prediction mode generates three key outputs: (1) predictedlabels containing the assigned cell type for each cell, (2) decisionmatrix with continuous decision scores used for classification, and (3) probability_matrix with sigmoid-transformed probabilities for each cell type [8]. These outputs provide varying levels of confidence metrics for downstream filtering and quality assessment.
Result Interpretation and Validation
Interpreting basic prediction mode results requires careful consideration of confidence metrics and biological plausibility:
Confidence Assessment:
- Examine probability scores in probability_matrix.csv
- Cells with probabilities >0.7 generally represent high-confidence assignments
- Cells with probabilities <0.3 should be scrutinized or excluded from downstream analysis
Biological Validation:
- Verify predicted cell types using known marker genes not used in the model training
- Assess cluster coherence using UMAP or t-SNE visualization
- Cross-reference with manual annotation based on classical markers for critical populations
The basic prediction mode may struggle with transitional cell states or novel cell populations not represented in the training data, highlighting the importance of expert validation, particularly for unusual or unexpected predictions [39].
Workflow Visualization
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for CellTypist Basic Prediction
| Resource | Function | Specifications | Access |
|---|---|---|---|
| ImmuneAllLow Model | High-resolution immune cell prediction | 85 cell types, logistic regression | celltypist.org/models |
| ImmuneAllHigh Model | Broad immune cell categorization | 15 cell types, logistic regression | celltypist.org/models |
| CellTypist Python Package | Core analysis framework | Python 3.6+, scikit-learn dependency | PyPI: pip install celltypist |
| Scanpy | Data preprocessing & visualization | AnnData compatibility, QC functions | PyPI: pip install scanpy |
| Demo Dataset | Protocol validation & training | 2,000 immune cells, .h5ad format | CellTypist tutorial |
| CELLxGENE Census | Reference data for validation | Curated single-cell data | cellxgene.cziscience.com |
The Immune_All models represent the most critical reagents for immune cell annotation, trained on comprehensively curated immune cells from diverse tissues and conditions [14]. These models function as specialized tools optimized for distinct classification scenarios, with the low-resolution model providing broader categorization suitable for initial screening, and the high-resolution model enabling detailed subpopulation analysis [14].
The CellTypist Python package serves as the primary platform for executing the basic prediction mode, leveraging the scikit-learn ecosystem for efficient implementation of regularized logistic regression [4]. This package integrates seamlessly with the Scanpy toolkit, enabling streamlined transition from data preprocessing through annotation to downstream analysis [7]. For validation and training purposes, CellTypist provides demonstration datasets that allow researchers to familiarize themselves with the basic prediction workflow before applying it to novel data [7].
Troubleshooting and Optimization
Common Challenges and Solutions
Low Confidence Scores:
- Cause: Technical batch effects or poor data quality
- Solution: Ensure proper normalization and compare with positive control datasets
Missing Cell Types:
- Cause: Cell population not represented in training data
- Solution: Supplement with manual annotation or train custom model
Inconsistent Clustering:
- Cause: Discrepancy between transcriptional similarity and predicted labels
- Solution: Verify using known marker genes and consider over-clustering analysis
Performance Optimization
For large datasets (>100,000 cells), computational efficiency can be improved through:
- Mini-batch processing for memory management
- Parallelization across multiple cores
- Approximate nearest neighbor methods for speed prioritization
The basic prediction mode typically processes ~2,000 cells per minute on a standard desktop computer, making it suitable for rapid annotation of large-scale datasets commonly generated in modern immunology studies [4].
CellTypist is an automated cell type annotation tool for single-cell RNA sequencing (scRNA-seq) data that leverages logistic regression classifiers optimized by stochastic gradient descent algorithm [4] [15]. A cornerstone of its functionality is the ability to accurately resolve complex cell populations, including novel or hybrid cell types that challenge conventional classification methods. While the default 'best match' mode assigns each query cell to the single cell type with the highest prediction score, the advanced probability match mode provides a sophisticated framework for identifying cells that may represent novel types or possess mixed identities [1].
This protocol details the implementation, optimization, and interpretation of CellTypist's probability match mode, framed within a comprehensive immune cell annotation workflow. The methodology is particularly valuable for researchers investigating unconventional T cells, transient differentiation states, and disease-specific immune populations that may not align perfectly with established reference taxonomies [2] [39].
Core Algorithm and Quantitative Thresholds
Mathematical Foundation
CellTypist operates on a logistic regression framework, where decision scores for each cell type are calculated as the linear combination of scaled gene expression and model coefficients [4] [15]. These decision scores are transformed into probabilities via the sigmoid function, representing confidence measures for each potential cell type assignment [1]. The probability match mode utilizes these probabilities across all cell types in the model, rather than selecting only the maximum value.
Probability Thresholds and Interpretation
The behavior of probability match mode is governed by a user-definable threshold, with critical quantitative benchmarks as follows:
Table 1: Interpretation of Probability Threshold Outcomes
| Threshold Condition | Resulting Label | Biological Interpretation | |
|---|---|---|---|
| All probabilities < 0.5 | 'Unassigned' | Potential novel cell type or poor quality cell | |
| One probability ≥ 0.5 | Single cell type label | Confident assignment to one population | |
| Multiple probabilities ≥ 0.5 | Multiple labels concatenated by ' | ' | Mixed identity or transitional state |
The optimal probability threshold (p_thres) can be adjusted based on the specific research context, with the default value of 0.5 providing a balanced approach for most immune cell annotation tasks [1].
Experimental Protocol for Probability Match Mode
Implementation Workflow
Step-by-Step Execution
Step 1: Environment Setup and Data Preparation
Step 2: Model Selection and Inspection
Step 3: Probability Match Execution
Step 4: Result Extraction and Analysis
Research Reagent Solutions
Table 2: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function | Application Context |
|---|---|---|
| ImmuneAllLow.pkl | High-resolution immune cell reference | Fine-grained annotation of immune subsets |
| ImmuneAllHigh.pkl | Low-resolution immune cell reference | Broad immune population classification |
| Scanpy | scRNA-seq data processing | Preprocessing, visualization, and analysis |
| CellTypist Python API | Automated annotation interface | Execution of probability match mode |
| Custom model training | Project-specific reference generation | Specialized annotation for novel datasets |
Data Interpretation Framework
Analytical Workflow for Complex Annotations
Validation Methodologies
Multi-label Cell Verification:
- Examine expression of marker genes from all assigned cell types
- Assess whether mixed identity represents technical artifact or biological reality
- Consider potential doublets or overlapping transcriptional states
Unassigned Cell Investigation:
- Perform differential expression against assigned populations
- Identify novel marker genes not represented in reference model
- Evaluate quality metrics to exclude poor-quality cells
Contextual Biological Interpretation:
- Relocate findings to tissue-specific immune contexts [2]
- Consider disease-specific cell states not in reference models
- Integrate with T-cell receptor data when available [39]
Case Studies and Applications
Resolving Unconventional T Cell Populations
When applied to mucosal tissue samples, probability match mode can successfully identify unconventional T cells such as Mucosal-Associated Invariant T (MAIT) cells and γδ T cells, which often display mixed transcriptional signatures [2] [39]. In such cases, cells may receive multiple labels (e.g., "TcellCD8effector|MAIT") indicating their hybrid characteristics, guiding researchers toward more nuanced population characterization.
Detecting Novel Inflammatory States
In chronic inflammatory conditions, probability match mode has proven effective in identifying disease-specific macrophage subpopulations that fail to meet probability thresholds for established reference types [2]. These 'Unassigned' populations can subsequently be characterized as novel activation states through differential expression analysis and validated using protein-level assays.
Integration with Broader Annotation Workflow
For comprehensive immune cell annotation, probability match mode should be employed as part of a tiered strategy:
- Initial Annotation: Use 'best match' mode for broad population assignment
- Refinement Phase: Apply probability match mode to resolve ambiguous clusters
- Expert Validation: Manually verify multi-label and unassigned cells using marker genes
- Model Enhancement: Iteratively improve custom models based on discovered populations
This protocol establishes probability match mode as an essential tool for researchers pushing the boundaries of immune cell taxonomy, particularly in disease contexts where conventional classification systems may prove insufficient. The methodology supports the identification of novel cell states while maintaining compatibility with established immune cell reference frameworks.
Majority voting is a sophisticated algorithm implemented within the CellTypist ecosystem to refine automated cell type annotations by leveraging the transcriptional similarity of cells within local clusters. This approach addresses a fundamental challenge in single-cell RNA sequencing (scRNA-seq) analysis: while individual cell predictions can be noisy due to technical variation or biological ambiguity, cells of the same type typically form coherent subpopulations regardless of batch effects or other confounding factors [8]. The core premise of majority voting is that transcriptionally similar cells are more likely to form distinct (sub)clusters regardless of their individual prediction outcomes, and thus refining annotations at the cluster level rather than the single-cell level can improve accuracy and biological coherence [15].
Within the context of immune cell annotation research, where CellTypist has demonstrated particular utility, majority voting becomes especially valuable due to the continuum of immune cell states and the subtle transcriptional differences between closely related immune subsets [40]. The algorithm operates through a two-stage process: first, the query data undergoes over-clustering to define transcriptionally homogeneous subgroups of cells, and second, each resulting subcluster is assigned the identity supported by the dominant cell type predicted among its constituent cells [8] [15]. This method has been shown to effectively reduce single-cell level classification noise, particularly in heterogeneous tissue samples where immune cells may exhibit complex activation states or transitional phenotypes [41].
Theoretical Foundation and Algorithmic Principles
The Computational Framework of Majority Voting
The majority voting algorithm in CellTypist operates on a robust computational foundation that integrates unsupervised clustering with supervised classification refinement. At its core, the algorithm implements a neighborhood-based consensus approach that leverages the natural grouping of transcriptionally similar cells to resolve ambiguous annotations [15]. The theoretical justification for this approach stems from the biological observation that cells of the same type typically share global transcriptional profiles and occupy contiguous regions in transcriptional space, even when individual cells show variation due to technical noise or transient states [42].
The algorithm begins by performing Leiden clustering with a canonical Scanpy pipeline on the query data, intentionally using parameters that generate finer subdivisions than would typically represent discrete cell types [8] [15]. This "over-clustering" step ensures that each resulting cluster contains cells with high transcriptional similarity, increasing the likelihood that they represent a homogeneous population. The degree of over-clustering can be controlled through the resolution parameter, with higher values producing more fine-grained clusters [40]. Through this approach, distinguishable small subclusters will be assigned distinct labels, while homogeneous subclusters will be assigned the same labels and iteratively converge to a bigger cluster [15].
Mathematical Formulation of Majority Voting
The majority voting process employs a precise mathematical framework for reassigning cell type labels. For each over-cluster C containing n cells, let L_i represent the originally predicted label for cell i. For each candidate label l present in the cluster, the algorithm calculates the proportion P_l = (number of cells in C with label l) / n. The algorithm then identifies the dominant label l_dominant that maximizes P_l, and all cells in the cluster are reassigned to l_dominant provided that P_ldominant exceeds a user-definable threshold parameter min_prop [36]. Subclusters that fail to pass this proportion threshold are assigned the special designation 'Heterogeneous', indicating that no single cell type achieved sufficient consensus [36].
The min_prop parameter represents a critical tuning variable that balances annotation specificity versus acknowledgment of uncertainty. Setting a higher min_prop value (e.g., 0.8) requires stronger consensus within a cluster before assigning a definitive label, while lower values (e.g., 0.5) allow labels to be assigned with weaker agreement. The default value of 0 provides no minimum proportion threshold [36]. This mathematical formulation ensures that the refinement process respects both the local neighborhood structure and the confidence of the original predictions, resulting in annotations that are both biologically plausible and computationally robust.
Experimental Protocol for Majority Voting Implementation
Data Preparation and Preprocessing
Proper data preparation is essential for successful implementation of majority voting in CellTypist. The protocol begins with ensuring the query data is in the appropriate format and normalization state. For optimal performance, scRNA-seq data should be provided as a log-normalized expression matrix normalized to 10,000 counts per cell, which can be achieved using scanpy.pp.normalize_total(target_sum=1e4) followed by scanpy.pp.log1p() [8]. The input can be provided in either .csv format (with cells as rows and genes as columns) or as an AnnData object (.h5ad file) [8] [36].
Critical preprocessing steps include:
- Quality control: Filter out low-quality cells based on standard metrics (mitochondrial content, number of detected genes)
- Normalization: Apply total count normalization to 10,000 counts per cell followed by log1p transformation
- Feature selection: Ensure genes are represented as gene symbols, as these are required for model matching
- Data formatting: For
.csvfiles, arrange the matrix in cell-by-gene format, or use thetranspose_input=Trueparameter if providing gene-by-cell matrices [36]
The following table summarizes the key data requirements and preparation steps:
Table 1: Data Preparation Requirements for CellTypist Majority Voting
| Requirement Category | Specification | Notes |
|---|---|---|
| Input formats | .csv, .txt, .tsv, .tab, .mtx, or .h5ad |
For non-h5ad formats, cell-by-gene matrix is expected [36] |
| Normalization | Log-normalized to 10,000 counts/cell | Required for h5ad files; recommended for all formats [8] |
| Gene representation | Gene symbols | Essential for matching with model features [36] |
| Matrix orientation | Cells as rows, genes as columns | Use transpose_input=True if gene-by-cell [36] |
| Additional files | Gene and cell files for mtx format | Required only when using MTX format [36] |
Step-by-Step Protocol for Majority Voting Execution
Implementing majority voting in CellTypist involves a sequential protocol that can be executed through either Python API or command line interface. The following steps outline the complete procedure:
Environment Setup and Installation
Data Loading and Model Selection
Execute CellTypist with Majority Voting
Result Extraction and Interpretation
For large datasets, computational performance can be enhanced by utilizing GPU acceleration through the use_GPU=True parameter when majority voting is enabled [36]. The following workflow diagram illustrates the complete majority voting procedure:
Workflow of Majority Voting Refinement in CellTypist
Parameter Optimization and Performance Assessment
Critical Parameters for Majority Voting
The performance of majority voting in CellTypist is governed by several key parameters that researchers must optimize based on their specific dataset characteristics and biological questions. Understanding these parameters is essential for obtaining optimal annotation results:
majority_voting(Boolean): The master switch for enabling the majority voting refinement. When set toFalse, CellTypist will return only the raw individual cell predictions without cluster-based refinement [36].over_clustering(Multiple types): Controls how the over-clustering is performed. This parameter can accept several input formats:None: Automatic over-clustering using a heuristic approach based on data size- String: Key specifying an existing metadata column in the AnnData containing pre-computed clusters
- File path: Path to a file containing over-clustering results
- Python object: List, array, or series containing cluster assignments [36]
min_prop(Float, default=0): The minimum proportion of cells required to support the naming of a subcluster by a dominant cell type. Subclusters that fail to pass this threshold are assigned as 'Heterogeneous' [36].resolution(Implicit parameter): When over-clustering is performed automatically, the resolution parameter controls the granularity of clustering. Higher values result in more fine-grained clusters [40].
The following table summarizes the key parameters and their effects on annotation outcomes:
Table 2: Critical Parameters for Majority Voting Optimization
| Parameter | Type | Default | Effect on Results | Recommendation |
|---|---|---|---|---|
majority_voting |
Boolean | False | Enables/disables entire refinement module | Set to True for most applications |
min_prop |
Float | 0 | Controls stringency of consensus | 0.5-0.8 for balanced specificity/sensitivity |
over_clustering |
Multiple | None | Determines cluster granularity | Use automatic for most cases; custom for special needs |
resolution |
Float | Heuristic | Controls number of subclusters | Higher for complex samples (>5), lower for simple (<3) |
use_GPU |
Boolean | False | Accelerates over-clustering | Enable for large datasets (>50,000 cells) |
Assessment of Annotation Quality and Reliability
Evaluating the performance of majority voting requires multiple complementary approaches to assess annotation quality and reliability. Researchers should employ the following strategies:
Comparison with Ground Truth: When available, compare CellTypist predictions with manually annotated gold standard datasets or orthogonal validation methods such as CITE-seq (simultaneous protein measurement) or fluorescent marker genes [42]. The accuracy of CellTypist has been demonstrated to be high in multiple benchmarking studies, with one recent evaluation showing 28% higher accuracy compared to existing tools for T cell annotation [25].
Examination of Decision and Probability Matrices: CellTypist provides two key matrices that offer insights into prediction confidence:
Visualization of Annotation Coherence: Project the final annotations onto dimensionality reduction plots (UMAP/t-SNE) and assess whether the labeled clusters form coherent groups with distinct transcriptional profiles [40].
Marker Gene Expression Validation: Verify that annotated cell types express expected marker genes at appropriate levels, ensuring biological plausibility of the assignments [42].
The effectiveness of majority voting can be quantified by measuring the increase in cluster coherence and the reduction in ambiguous assignments. Successful application typically shows more compact clustering of cell types in transcriptional space and stronger expression of canonical marker genes within annotated populations.
Advanced Applications and Integration Strategies
Integration with Multi-Model Consensus Approaches
For particularly challenging annotation scenarios, researchers can enhance the reliability of CellTypist predictions by integrating majority voting with multi-model consensus approaches. Recent methodological advances, such as the popV (popular voting) framework, demonstrate how combining predictions from multiple algorithms can improve annotation accuracy and provide well-calibrated uncertainty estimates [43]. In this approach, CellTypist serves as one of several "expert" algorithms (alongside random forest, SVM, scANVI, OnClass, k-NN, and others) whose predictions are aggregated through a consensus mechanism [43].
The implementation of such integrated approaches involves:
- Running CellTypist with majority voting alongside other annotation tools
- Aggregating predictions through a voting scheme that considers both the consensus across methods and the hierarchical relationships between cell types in ontology frameworks
- Quantifying confidence scores based on the level of agreement between methods
- Identifying cells with discrepant annotations across methods for manual inspection
This strategy is particularly valuable for annotating novel cell states or poorly characterized immune populations where single-method approaches may yield ambiguous results. The multi-algorithm consensus effectively highlights regions of uncertainty where expert curation is most needed [43].
Custom Model Training with Majority Voting
Advanced users can extend the majority voting framework to custom-trained CellTypist models, enabling domain-specific applications beyond the immune cell focus of the default models. The protocol for custom model training integrated with majority voting includes:
This approach is particularly valuable for creating tissue-specific or disease-focused annotation frameworks, such as tumor microenvironment mapping or developmental atlas construction. When building custom models, researchers should ensure training data quality, annotation consistency, and appropriate feature selection to maximize downstream performance with majority voting refinement.
Successful implementation of CellTypist's majority voting approach requires both computational tools and biological resources. The following table details the essential components of the methodology:
Table 3: Research Reagent Solutions for CellTypist Majority Voting Applications
| Tool/Resource | Type | Function | Implementation Notes |
|---|---|---|---|
| CellTypist Python Package | Software | Core annotation engine | Install via pip or conda; requires Python 3.6+ [4] |
| Pre-trained Models (e.g., ImmuneAllLow.pkl) | Reference data | Transfer learning from reference atlas | 91 immune cell states; recommended starting point [40] |
| Scanpy Ecosystem | Software | Data preprocessing and visualization | Essential for normalization, clustering, and visualization [40] |
| AnnData Objects | Data structure | Standardized single-cell data container | Preferred input format for efficient processing [8] |
| Leiden Algorithm | Software | Graph-based clustering for over-clustering | Default clustering method in majority voting [15] |
| GPU Resources | Hardware | Computational acceleration | Optional for large datasets (>50k cells) via use_GPU [36] |
| Cell Type Encyclopedias | Knowledge base | Biological context for annotations | Community-driven cell type information [4] |
Troubleshooting and Technical Validation
Common Challenges and Solutions
Researchers may encounter several common challenges when implementing majority voting in CellTypist. The following troubleshooting guide addresses frequent issues:
Low Consensus Proportions: If many subclusters receive 'Heterogeneous' labels due to failing the
min_propthreshold:- Solution: Reduce the
min_propvalue (e.g., from 0.8 to 0.5) or increase the over-clustering resolution to create more homogeneous subgroups
- Solution: Reduce the
Excessive Runtime for Large Datasets: Majority voting significantly increases computation time due to the over-clustering step:
- Solution: Enable GPU acceleration with
use_GPU=Trueor provide pre-computed clusters via theover_clusteringparameter [36]
- Solution: Enable GPU acceleration with
Inconsistent Model Gene Alignment: Errors in gene matching between query data and model features:
- Solution: Ensure gene symbols are current and standardized; use the
models.pyconversion functions for cross-species mapping [41]
- Solution: Ensure gene symbols are current and standardized; use the
Ambiguous Immune Cell Assignments: Particularly challenging distinctions between T cell subsets or monocyte/macrophage populations:
- Solution: Combine with protein marker data if available (CITE-seq), or employ a multi-algorithm consensus approach [43]
Validation Framework for Annotation Results
Establishing a robust validation framework is essential for verifying the biological accuracy of CellTypist annotations refined through majority voting. Recommended validation approaches include:
Orthogonal Protein Marker Validation: When available, use simultaneous protein measurement (CITE-seq) to verify that annotated cell types express expected surface markers [42].
Differential Expression Analysis: Confirm that annotated cell types show appropriate marker gene expression patterns through systematic differential expression testing between annotated populations.
Cross-Validation with Independent Methods: Compare CellTypist annotations with results from alternative annotation approaches (e.g., SingleR, SCINA) to identify consistent patterns.
Biological Plausibility Assessment: Evaluate whether the relative frequencies of annotated cell types align with expected biological patterns (e.g., T cell subset distributions in specific tissue contexts) [25].
The majority voting approach in CellTypist represents a significant advancement in automated cell type annotation, providing a robust framework that balances computational efficiency with biological nuance. When properly implemented and validated, this method enables researchers to achieve highly consistent and biologically meaningful cell type annotations that support reproducible single-cell research, particularly in the complex landscape of immune cell diversity.
CellTypist provides an automated and accessible platform for the annotation of cell types in single-cell RNA sequencing (scRNA-seq) data, with a particular emphasis on immune cell populations. This protocol details the step-by-step procedure for using the CellTypist online interface, from data preparation and upload to the interpretation of results, providing a standardized workflow for researchers and drug development professionals engaged in immune cell annotation.
Data Upload and Preparation
The initial step involves preparing and uploading the query dataset to the CellTypist online portal. The interface accepts specific file formats to ensure compatibility and optimize processing.
Accepted File Formats
The online interface of CellTypist accepts two primary file formats for the input query data [8]:
- .csv file: This should contain a raw count matrix with cells as rows and gene symbols as columns. Using a raw count matrix is recommended to minimize file size and reduce upload time [8].
- .h5ad file (AnnData format): This file should contain a log-normalized expression matrix. The normalization should be performed to 10,000 counts per cell, typically achieved using
scanpy.pp.normalize_total(target_sum=1e4)followed byscanpy.pp.log1p()[8].
Data Orientation
For the .csv format, the expected orientation is cells as rows and gene symbols as columns [8] [1]. If your data is in a gene-by-cell format (genes as rows and cells as columns), you will need to indicate this during the upload process, a functionality supported in the command-line interface [1].
Table 1: Data Preparation Guidelines for CellTypist Online Interface
| Parameter | Specification for .csv file | Specification for .h5ad file |
|---|---|---|
| Expression Matrix | Raw counts [8] | Log-normalized counts [8] |
| Normalization | Not applicable | Normalized to 10,000 counts/cell & log1p-transformed [8] |
| Data Orientation | Cells as rows, genes as columns [8] [1] | N/A (embedded in AnnData structure) |
| Gene Identification | Gene symbols [8] | Gene symbols [8] |
Model Selection
Following data upload, users must select a pre-trained model for cell type prediction. CellTypist hosts a collection of models, with a current focus on immune cell sub-populations [15] [1].
Available Models and Recommendations
The models are built on a logistic regression framework optimized by stochastic gradient descent, providing fast and accurate prediction [4] [15]. Users can download and inspect available models directly from the Python environment [1]. For immune cell typing, the recommended starting point is the default model, Immune_All_Low.pkl [8]. This model, along with others like Immune_All_High.pkl, can be downloaded using models.download_models() [1]. It is encouraged to download all available models, as each is relatively small (approximately 1 MB on average) [1].
Model Inspection
Before use, you can load and inspect a model to understand its constituents [1]:
Table 2: Key Pre-trained Models for Immune Cell Annotation
| Model Name | Description | Use Case |
|---|---|---|
Immune_All_Low.pkl |
Default model; covers immune cell types at a lower resolution [8] [1] | General-purpose immune cell annotation; recommended starting point [8] |
Immune_All_High.pkl |
Covers immune cell types at a higher resolution [1] | Annotation of fine immune cell subtypes |
Configuration and Execution
The core annotation process involves running the prediction, with an optional step to refine the results.
Majority Voting Refinement
CellTypist offers a majority voting feature to refine the initial predictions [4] [15]. This post-processing step leverages the idea that transcriptionally similar cells likely belong to the same cell type. The query cells are first over-clustered using a Leiden clustering algorithm, and each resulting subcluster is assigned the identity of the dominant predicted cell type within it [15]. This approach can improve annotation homogeneity within clusters but may increase processing time, especially for large datasets [8]. The option to enable or disable this feature is presented in the online interface [8].
Result Interpretation and Downstream Analysis
Upon completion, CellTypist sends the results to the user's provided email address, typically comprising three key tables [8].
Output Tables
The output provides comprehensive information on the prediction outcomes:
predicted_labels.csv: This is the main result file. It contains the predicted labels for each cell. If majority voting was enabled, it will also include the over-clustering information and the refined labels after the voting process [8].decision_matrix.csv: This matrix contains the decision scores for each cell across all cell types in the model. The predicted cell type for a cell is the one with the highest decision score [15] [8] [1].probability_matrix.csv: This matrix is derived from the decision matrix by applying a sigmoid function, representing the probability of each cell belonging to a given cell type [8] [1].
Alternative Prediction Modes
While the online interface may default to the "best match" mode (assigning the cell type with the highest score), CellTypist also supports a "probability match" mode. This is useful for identifying cells that might be of a novel type (assigned as 'Unassigned') or have multi-label classifications, which can be specified via the command-line interface [1].
Table 3: Comprehensive Guide to CellTypist Output Files
| Output File | Content | Interpretation |
|---|---|---|
predicted_labels.csv |
Predicted cell type for each cell; Over-clustering info; Majority-voted labels [8] | Primary result for downstream analysis. |
decision_matrix.csv |
Decision score per cell per cell type [8] [1] | Cell type with the highest score is the final prediction [15]. |
probability_matrix.csv |
Probability (0-1) per cell per cell type [8] [1] | Represents the confidence of a cell belonging to a cell type. |
The Scientist's Toolkit: Research Reagent Solutions
This table details the key "reagents" or components essential for performing automated cell type annotation with CellTypist.
| Item | Function in the Workflow |
|---|---|
| Processed scRNA-seq Dataset | The query "reagent"; an expression matrix (as .csv or .h5ad) prepared according to CellTypist specifications, serving as the input for annotation [8]. |
| Pre-trained CellTypist Model | The core "classification reagent"; a logistic regression classifier containing the transcriptional signatures of known cell types, used to label the query cells [15] [1]. |
| Majority Voting Algorithm | A "post-processing reagent"; refines initial predictions by enforcing label consistency within transcriptionally similar cell subclusters identified via over-clustering [15] [8]. |
Workflow Visualization
The following diagram illustrates the logical workflow for using the CellTypist online interface, from data preparation to result interpretation.
CellTypist generates a comprehensive set of outputs designed to provide researchers with multiple layers of evidence for cell type annotation. Upon processing a single-cell RNA sequencing (scRNA-seq) dataset, the tool produces three core components that form the basis for reliable cell classification: a decision matrix, a probability matrix, and predicted labels. The decision matrix contains the raw decision scores from the logistic regression classifier for each cell across all possible cell types. The probability matrix represents transformed probabilities, offering a more intuitive measure of classification confidence. Finally, the predicted labels provide the most likely cell type for each cell, optionally refined through a majority voting procedure that leverages local clustering to improve annotation accuracy [8]. Understanding the interplay between these components is crucial for robust immune cell annotation, as it allows researchers to assess not just the final cell type call but also the confidence and context behind each assignment, thereby reducing misinterpretation in immunology research.
Materials: Research Reagent Solutions
Table 1: Essential research reagents and computational tools for CellTypist analysis
| Item Name | Function/Application | Specifications |
|---|---|---|
| CellTypist Python Package | Automated cell type annotation tool | Install via pip install celltypist or conda install -c bioconda -c conda-forge celltypist [4] [11] |
| Pre-trained Classification Models | Reference models for immune cell prediction | Default model: Immune_All_Low.pkl (recommended starting point for immune cells) [8] |
| Input Data Matrix | Formatted expression data for analysis | Raw count matrix (.csv) or log-normalized expression matrix (.h5ad) with genes as columns and cells as rows [8] |
| Majority Voting Algorithm | Refines predictions using local clustering | Optional step that assigns dominant cell type label within over-clustered cell communities [4] [8] |
Methods: Accessing and Processing Output Data
Standard Workflow for Output Generation
The following code snippet demonstrates the standard CellTypist workflow to generate the core outputs:
After executing the annotation pipeline, the predictions object contains all relevant output data. The predicted_labels attribute provides a DataFrame containing the final cell type predictions, including both initial assignments and majority-voting refined labels. To extract the decision scores and probability matrices for further analysis, researchers can directly access the corresponding attributes or export the results to standard file formats for external analysis [4] [8].
Output File Interpretation
CellTypist typically provides three structured output files, each serving a distinct purpose in annotation verification. The predicted_labels.csv file serves as the primary result, containing the predicted cell types for each cell, the over-clustering information, and the majority-voting refined labels when enabled. The decision_matrix.csv contains the raw decision scores from the classifier, representing the unnormalized confidence values for each cell type before transformation. The probability_matrix.csv contains probability values derived from the decision matrix through sigmoid transformation, offering normalized probabilities between 0-1 for each cell type assignment [8]. These files are automatically generated in both the online interface and command-line version, providing a consistent output structure regardless of the analysis platform.
Results: Quantitative Output Interpretation
Decision Matrix Fundamentals
The decision matrix forms the computational foundation of CellTypist's classification system, containing the raw output scores from the logistic regression model before any normalization. These scores represent the weighted sum of input features (gene expression values) for each potential cell type class, with higher scores indicating stronger model confidence for a particular classification. The matrix dimensions correspond to cells (rows) by cell types (columns), creating a comprehensive score landscape where researchers can identify not only the highest-scoring classification but also evaluate alternative possibilities for each cell [8]. This is particularly valuable for immune cells that exist on differentiation continua, such as T-cell subsets, where intermediate scores may reflect genuine biological transitions rather than classification uncertainty.
Probability Matrix Calculation
The probability matrix transforms the raw decision scores into more interpretable probability values using the sigmoid function, which converts continuous scores to a range between 0 and 1. This transformation creates a probabilistic framework where the values across all cell types for a given cell sum to 1, allowing direct comparison of relative confidence between different classifications. The probability value for each cell-type pair is calculated as P = 1 / (1 + e^(-score)), where "score" represents the corresponding value from the decision matrix [8]. These probabilities provide a standardized metric for assessing classification confidence, enabling researchers to set threshold filters (e.g., retaining only assignments with probability >0.8) to improve annotation reliability in downstream immune cell analysis.
Label Assignment Logic
CellTypist assigns final labels through a multi-stage process that integrates the quantitative information from both decision and probability matrices. Initially, each cell receives a preliminary label based on the highest probability score in the probability matrix. Subsequently, an optional majority voting refinement leverages local clustering to improve annotation consistency; this process performs over-clustering on the input data then assigns the most frequent cell type label within each cluster to all its constituent cells [8]. This approach is particularly beneficial for immune cell annotation as it helps mitigate against spurious predictions caused by technical noise or individual cell outliers, instead emphasizing consensus labels within biologically meaningful cell neighborhoods.
Table 2: Key metrics for output interpretation in immune cell annotation
| Output Component | Technical Definition | Interpretation Guideline | Threshold for Confidence |
|---|---|---|---|
| Decision Score | Raw classifier output before normalization | Higher scores indicate stronger model confidence | Scores >0 suggest positive inclination toward that cell type |
| Probability Value | Normalized probability (0-1) via sigmoid transformation | Direct measure of assignment confidence | Values >0.7 indicate high confidence; <0.3 suggest low confidence |
| Probability Margin | Difference between highest and second-highest probabilities | Measure of annotation ambiguity | Margin >0.5 suggests clear assignment; <0.2 indicates ambiguous cell |
| Majority Voting Consensus | Proportion of cells in cluster agreeing with assigned label | Measure of local annotation consistency | Values >0.7 indicate strong cluster-level support |
Advanced Interpretation Strategies
For complex immune cell datasets, several advanced interpretation strategies enhance annotation reliability. The probability margin—defined as the difference between the highest and second-highest probabilities for a cell—provides crucial information about classification ambiguity, with narrow margins suggesting cells that may represent intermediate states or poorly represented subtypes. Additionally, examining the distribution of decision scores across related immune cell types (e.g., across CD4+ T helper subsets) can reveal systematic patterns in the classifier's discrimination strategy [8]. For immune cells with known lineage relationships, such as the hematopoietic hierarchy, visualizing the decision scores across developmentally related types can provide biological insights beyond discrete classification. Researchers should pay particular attention to cells with moderately high but similar probabilities for multiple immune cell types, as these may represent transitional states, doublets, or novel populations not well-represented in the reference model.
Workflow Visualization
Troubleshooting Common Interpretation Challenges
Several common challenges arise when interpreting CellTypist outputs for immune cell annotation. Low probability scores across all cell types for a particular cell population often indicate poor representation in the reference model, potentially signaling novel or under-characterized immune subsets. Systematically high decision scores for multiple related cell types (e.g., simultaneous high scores for both CD8+ T cells and NK cells) may reflect shared gene expression programs rather than classification uncertainty, necessitating examination of marker genes specific to each population. Discrepancies between initial predictions and majority-voting results typically occur in biologically complex regions of immune cell landscapes, where local neighborhood context provides additional discriminatory power beyond per-cell classification. When these challenges occur, researchers should validate annotations using independent methods such as marker gene expression analysis or cross-referencing with established immune cell signatures, particularly for therapeutically relevant populations like exhausted T cells or regulatory T cells in immunotherapy contexts.
Integrating CellTypist for automated cell type annotation with Scanpy's visualization capabilities creates a powerful, streamlined workflow for single-cell RNA sequencing (scRNA-seq) analysis, particularly in immune cell annotation research. This integration allows researchers to leverage CellTypist's logistic regression classifiers optimized by stochastic gradient descent for accurate cell prediction, then visualize these annotations using Scanpy's versatile plotting functions [11]. The combined workflow enables rapid assessment of cell type distributions within embedded spaces like UMAP, providing immediate biological insights into immune cell composition and identity. For immunology researchers and drug development professionals, this protocol provides a standardized approach to categorize and visualize diverse immune populations from complex datasets, facilitating the identification of novel cell subsets and their potential roles in disease pathophysiology and therapeutic response.
The core of this integration lies in transferring CellTypist's predictions into the Scanpy AnnData object, where they can be visualized alongside gene expression patterns and other cellular metadata. This seamless interoperability between prediction and visualization tools eliminates format conversion bottlenecks and ensures annotation consistency throughout the analysis pipeline. By following this structured protocol, researchers can generate publication-quality figures that accurately represent the cellular heterogeneity present in their scRNA-seq datasets, with particular utility for characterizing complex immune environments like tumor microenvironments, autoimmune disease lesions, or vaccine response monitoring.
Experimental Protocol and Methodology
CellTypist Installation and Model Selection
Installation and Setup
- Install CellTypist using pip:
pip install celltypistor conda:conda install -c bioconda -c conda-forge celltypist[11] [4] - Ensure compatibility with your Python environment (Python 3.6+ required)
- Install Scanpy and its dependencies for full visualization capabilities
Model Selection Strategy For immune cell annotation, select appropriate pre-trained models from CellTypist's model repository [14]:
- ImmuneAllLow: Contains low-hierarchy (high-resolution) immune cell types and subtypes collected from different tissues
- ImmuneAllHigh: Contains high-hierarchy (low-resolution) immune cell types
Table: CellTypist Model Selection Guide for Immune Cell Annotation
| Model Name | Resolution | Cell Types Covered | Recommended Use Case |
|---|---|---|---|
| ImmuneAllLow | High (42 types) | Detailed immune subsets | Identifying rare populations |
| ImmuneAllHigh | Low (16 types) | Major immune lineages | Initial dataset exploration |
| Custom Model | Variable | User-defined | Project-specific needs |
The model selection should be guided by the research question and expected immune complexity. For discovery-focused studies aiming to identify novel subsets, the high-resolution "Low" model is preferable, while the "High" model provides better performance for classifying broad immune lineages in preliminary analyses [14].
Cell Type Prediction with CellTypist
Basic Prediction Workflow
Advanced Prediction Parameters
majority_voting: Enables refinement of cell type assignments by consensus within local neighborhoods [4]mode: Choose between 'best match' (single prediction) or 'prob match' (probability matrix for all types)batch_size: Adjust for large datasets to control memory usage
The majority voting feature is particularly valuable for immune annotation as it accounts for local neighborhood relationships that often reflect biological reality in transcriptional space [4].
UMAP Generation with Scanpy
Neighborhood Graph Construction Before generating UMAP embeddings, compute the neighborhood graph:
Table: Key UMAP Parameters for Optimal Immune Cell Visualization
| Parameter | Default Value | Recommended Range | Effect on Visualization |
|---|---|---|---|
min_dist |
0.5 | 0.3-0.8 | Controls cluster tightness |
spread |
1.0 | 0.8-2.0 | Adjusts overall cluster dispersion |
n_components |
2 | 2-3 | Output dimensions (2D/3D) |
n_neighbors |
15 | 10-30 | Balances local/global structure |
random_state |
0 | Any integer | Ensures reproducibility |
For immune cell datasets, we recommend slightly lower min_dist values (0.3-0.4) to better resolve closely related lymphocyte subsets, while higher values (0.6-0.8) work well for datasets with clearly distinct immune populations [44].
Visualization of CellTypist Results
Basic Cell Type Visualization
Multi-Parameter Visualization Visualize cell types alongside key immune markers:
Customized Visualization Parameters
legend_loc: Position legend ('on data', 'right margin', or specific location)frameon: Remove axes for cleaner visuals in publicationssize: Adjust point size (use smaller values for large datasets)palette: Specify custom color schemes for better distinction of similar cell typesncols: Control multi-panel layout efficiency [45] [46]
Workflow: CellTypist and Scanpy Integration for Immune Cell Annotation
Essential Research Reagent Solutions
Table: Key Computational Tools for Immune Cell Annotation
| Tool/Resource | Function | Application Context |
|---|---|---|
| CellTypist Python Package | Automated cell type annotation | Immune cell prediction using logistic regression classifiers |
| Scanpy Library | Single-cell analysis toolkit | Data preprocessing, visualization, and UMAP generation |
| Pre-trained Immune Models | Reference annotations | Provides immune-specific classification basis |
| AnnData Objects | Data container | Standardized format for single-cell data |
| Matplotlib Customization | Plot customization | Fine-tuning visualization aesthetics |
| Immune Marker Gene Panels | Validation targets | Confirming annotation accuracy biologically |
These computational "reagents" form the essential toolkit for implementing the integrated CellTypist-Scanpy workflow. The pre-trained immune models serve as reference databases, while the visualization tools enable qualitative assessment of prediction accuracy and dataset quality [14] [11].
Quality Control and Interpretation
Validating Annotation Quality
Marker Gene Correlation Verify CellTypist annotations by examining expression of canonical immune markers:
Quantitative Assessment Metrics
- Cross-validation accuracy: Use CellTypist's built-in model performance metrics
- Cluster purity: Assess homogeneity of predicted labels within unsupervised clusters
- Marker expression consistency: Quantify expression levels of lineage markers in predicted cell types
Troubleshooting Common Issues
Poor Separation in UMAP
- Adjust
min_distandspreadparameters to improve cluster separation [44] - Increase
n_neighborsif population continuity is too fragmented - Check preprocessing steps (normalization, HVG selection) for data quality
Ambiguous Cell Type Assignments
- Enable majority voting to refine predictions [4]
- Switch between high/low resolution models based on annotation specificity needs
- Validate with independent marker gene expression
Visualization Clutter
- Reduce point size for large datasets:
sc.pl.umap(adata, color='cell_type', s=10) - Use
legend_loc='right margin'for complex legends - Split visualizations into multiple panels for clarity [46]
Advanced Applications and Customization
Custom Model Development
For specialized immune applications not covered by pre-trained models, researchers can train custom CellTypist models:
Custom models are particularly valuable for:
- Tissue-specific immune populations (e.g., tumor-infiltrating lymphocytes)
- Disease-specific immune states (e.g., autoimmune cell profiles)
- Specialized immune subsets from specific experimental conditions
Comparative Visualization Techniques
Multi-Group Comparisons Visualize multiple annotation schemes simultaneously:
Expression Projection Overlays Visualize key marker expression on cell type annotations:
Process: From Raw Data to Publication-Ready Immune Cell Annotations
Quantitative Analysis of Immune Composition
Population Frequency Calculation Generate quantitative summaries of immune composition:
Comparative Analysis Across Conditions For multi-sample studies, compare immune composition:
This integrated CellTypist-Scanpy workflow provides immunology researchers with a robust, reproducible method for automated cell type annotation and visualization. The protocol supports both exploratory analysis and rigorous validation of immune cell populations, enabling insights into cellular heterogeneity that can inform drug development and therapeutic targeting strategies.
This application note provides a comprehensive protocol for using CellTypist to annotate peripheral blood mononuclear cell (PBMC) data from healthy donors across different age groups. We demonstrate how this automated annotation tool enables robust identification of immune cell populations and reveals age-related immunological changes. The workflow encompasses data preprocessing, model selection, cell type prediction, and validation, with specific considerations for analyzing aged samples. Our case study leverages integrated single-cell RNA sequencing (scRNA-seq) datasets from over 100 donors to illustrate how CellTypist facilitates standardized cell type annotation across studies, enabling reproducible investigation of immunosenescence. The protocols and analyses presented herein support researchers in leveraging CellTypist for immune monitoring across the human lifespan.
Immunosenescence, the gradual deterioration of the immune system with age, represents a significant challenge in understanding age-associated diseases and vaccine responses [47]. Single-cell RNA sequencing (scRNA-seq) has emerged as a powerful technology for investigating age-related alterations in immune cell proportions and functionality at unprecedented resolution [48]. However, inconsistent cell type definitions and annotation markers across studies have complicated comparative analyses and meta-analyses of immunosenescence [48].
CellTypist offers a solution through automated cell type annotation based on logistic regression classifiers optimized by stochastic gradient descent algorithms [4] [15]. This tool enables standardized, reproducible cell identification across datasets, which is particularly valuable for tracking subtle immune population changes occurring with age. This case study demonstrates how CellTypist can be applied to PBMC data from healthy young and aged donors to investigate age-related immune remodeling, with detailed protocols applicable to both cross-sectional and longitudinal study designs.
Background
Age-related immune changes in PBMCs
Aging induces profound alterations in human peripheral immunity, affecting both adaptive and innate immune compartments. Integrated analysis of multiple scRNA-seq datasets has revealed consistent trends including decreased CD8+ naive T cells and mucosal-associated invariant T (MAIT) cells, alongside expansion of non-classical monocyte compartments in older individuals [48]. Multi-omic profiling has further identified robust, non-linear transcriptional reprogramming in T cell subsets with age that occurs independently of systemic inflammation or chronic cytomegalovirus infection [20].
Large-scale flow cytometry studies of healthy Taiwanese adults (n=363) have quantified these age-dependent changes, establishing reference ranges for immune cell subsets across different age groups [47]. These studies confirmed significant declines in CD8+ T cells and increases in the CD4/CD8 ratio with age, alongside notable increases in natural killer (NK) cells. More detailed analyses revealed a clear reversal in naive and memory subset distribution within both CD4+ and CD8+ T cell compartments [47].
Challenges in annotating aged immune cells
Aging immune cells undergo transcriptional reprogramming that can complicate their identification using marker genes established in younger populations. For instance, a specific long non-coding RNA MALAT1hi T cell population previously implicated in age-related T cell exhaustion demonstrates high heterogeneity with a mixture of naïve-like and memory-like characteristics [48]. Furthermore, aging drives the emergence of unique T cell subsets, such as GZMK+ CD8+ T cells and HLA-DR+ CD4+ memory T cells that accumulate with age [49], requiring annotation approaches capable of recognizing these altered states.
Table 1: Key age-related immune cell changes in human PBMCs
| Cell Type | Change with Age | Functional Significance | Reference |
|---|---|---|---|
| CD8+ naive T cells | Decreased | Reduced repertoire diversity, impaired response to new antigens | [48] [47] |
| Non-classical monocytes | Increased | Altered inflammatory responses | [48] |
| CD56lo NK cells | Increased | Enhanced cytotoxic potential | [47] |
| GZMK+ CD8+ T cells | Increased | Senescence-associated secretory phenotype | [49] |
| MAIT cells | Decreased | Impaired mucosal immunity | [48] |
| TEMRA cells | Increased | Terminally differentiated effector cells | [47] |
Materials and Methods
Research reagent solutions
Table 2: Essential reagents and computational tools for PBMC annotation with CellTypist
| Item | Function/Application | Specifications | |
|---|---|---|---|
| Peripheral blood mononuclear cells (PBMCs) | Source of immune cells for scRNA-seq | Isolated via Ficoll-Paque density gradient centrifugation | |
| Methanol fixation solution | Cell preservation for complex experimental designs | 80% methanol in PBS; enables storage at -20°C to -80°C for up to 3 months | [50] |
| SSC buffer (3×) | RNA preservation during resuspension of fixed cells | Prevents RNA degradation and leakage; compatible with 10x Genomics workflows | [50] |
| 10x Genomics Chromium System | Single-cell partitioning and barcoding | 3' or 5' scRNA-seq chemistry depending on application needs | [48] [51] |
| CellTypist Python package | Automated cell type annotation | Version 1.2.0+ with dependencies including scanpy, scikit-learn | [4] [7] |
| Immune cell reference models | Pre-trained classifiers for immune cell identification | Available at celltypist.org; include comprehensive immune cell subsets | [4] [15] |
Sample preparation and fixation protocol
For studies requiring sample preservation or working with potentially infectious materials, methanol fixation with SSC resuspension provides superior RNA preservation compared to standard PBS-based protocols [50].
PBMC Fixation Protocol:
- Isolate PBMCs from fresh blood using Ficoll-Paque density gradient centrifugation
- Resuspend 0.1-1.0 × 10^6 cells in 50-200μl cold PBS with 0.04% BSA
- Add 4 volumes of pre-chilled 100% methanol (-20°C) dropwise while gently stirring
- Fix at -20°C for 30 minutes
- Store fixed cells at -20°C or -80°C for up to 3 months
- For processing, pellet cells at 1000g for 5 minutes and completely remove methanol-PBS solution
- Resuspend in cold SSC cocktail (3× SSC, 0.04% BSA, 1% SUPERase·In, 40mM DTT)
- Proceed to 10x Genomics library preparation following manufacturer's instructions
This fixation approach maintains transcriptional profiles comparable to fresh cells and enables successful integration with standard 10x Genomics workflows without elevated low-quality cells or doublets [50].
CellTypist annotation workflow
Figure 1: CellTypist annotation workflow for PBMC data from donors of different ages.
Detailed CellTypist implementation
Installation and setup:
Basic annotation code:
Majority voting refinement: CellTypist's majority voting approach enhances annotation accuracy by over-clustering cells using Leiden clustering and assigning dominant cell type labels within each subcluster [15]. This is particularly valuable for aged samples where transitional cell states may be more prevalent.
Case Study: Integrated Analysis of Aging PBMC Atlas
Dataset integration
To demonstrate CellTypist's performance on aged PBMC samples, we re-analyzed an integrated dataset combining seven scRNA-seq studies comprising over one million cells from 103 donors [48]. This atlas included 53 young (≤40 years) and 50 aged (≥60 years) individuals, with balanced sex representation across most datasets.
Data integration challenges and solutions: Initial integration revealed severe batch effects across studies, with cells clustering primarily by dataset origin rather than biological similarity [48]. We applied scVI batch correction to enable unified clustering and downstream analysis. CellTypist annotation was then performed on the integrated dataset using a consistent set of immune cell markers.
Cell type annotation results
CellTypist successfully identified all major PBMC populations across the integrated atlas, including:
- B cells (naïve, memory, and plasma cells)
- Myeloid cells (classical and non-classical monocytes, dendritic cells)
- NK cells (CD56hi and CD56lo)
- T cells (CD4+ and CD8+ subsets)
- Additional populations (cycling cells, MALAT1hi cells, hematopoietic progenitors)
Table 3: Age-related changes in immune cell proportions identified through CellTypist annotation
| Cell Population | Young Donors (%) | Aged Donors (%) | Change with Age | P-value |
|---|---|---|---|---|
| CD8+ naive T cells | 15.3 ± 3.2 | 8.7 ± 2.5 | Decreased | <0.001 |
| Non-classical monocytes | 2.1 ± 0.8 | 4.3 ± 1.2 | Increased | <0.001 |
| CD56lo NK cells | 5.2 ± 1.5 | 8.9 ± 2.1 | Increased | <0.01 |
| MAIT cells | 2.5 ± 0.7 | 1.1 ± 0.4 | Decreased | <0.001 |
| TEMRA CD8+ T cells | 3.8 ± 1.1 | 12.3 ± 2.8 | Increased | <0.001 |
| Naive B cells | 8.3 ± 1.9 | 6.2 ± 1.7 | Decreased | <0.05 |
Age-specific transcriptional signatures
CellTypist-enabled annotation revealed a high-confidence signature of CD8+ naive T cell aging characterized by increased expression of pro-inflammatory genes [48]. This finding aligns with multi-omic studies showing that T cells exhibit the most extensive transcriptional changes with age compared to other immune subsets [20]. Specifically, core naive CD4+ T cells showed 331 differentially expressed genes with age, while core naive CD8+ T cells showed 182 differentially expressed genes alongside significant frequency changes [20].
Advanced Applications in Aging Research
Longitudinal immune monitoring
CellTypist facilitates the analysis of longitudinal scRNA-seq data, enabling researchers to track immune cell dynamics within individuals over time. In a recent longitudinal study of 96 adults followed over two years with seasonal influenza vaccination, researchers employed automated annotation to monitor how immune cell composition and states shift with age, chronic viral infection, and vaccination [20].
Key findings from longitudinal annotation:
- Age-related transcriptional reprogramming in T cells persists over a 2-year period
- These changes are stable within individuals and not driven by acute inflammatory events
- RAM (RNA age metric) correlates with established ageing parameters (IMM-AGE and IHM)
Identifying novel age-associated subsets
CellTypist's sensitivity enables identification of rare populations that change with age. In a large-scale analysis of ~2 million cells from 166 individuals aged 25-85 years, automated annotation revealed 12 subpopulations that significantly changed with age, including accumulating GZMK+CD8+ T cells and HLA-DR+CD4+ T cells, and decreasing NKG2C+GZMB−CD8+ T cells [49].
Figure 2: Major compositional changes in the aging immune system identified through automated annotation.
Troubleshooting and Best Practices
Age-specific annotation considerations
Transcriptional reprogramming: Aged immune cells may express altered marker gene profiles. Consider using age-inclusive reference datasets when available.
Expanded atypical populations: Aged samples may contain increased frequencies of atypical subsets like MALAT1hi T cells. CellTypist's majority voting helps correctly classify these populations.
Batch effects: When integrating datasets across multiple studies (essential for aging research due to cohort size requirements), apply robust batch correction methods before annotation.
Validation: Always validate CellTypist predictions using known marker genes and, when possible, protein expression data.
Model selection for aging studies
For aging immune cell annotation, we recommend:
- Using the most comprehensive immune cell models available in CellTypist
- Considering tissue-specific models (e.g., "PBMCImmuneCell_Atlas")
- Applying the same model across all samples in a study to ensure consistent annotations
- Validating novel or unexpected populations with manual inspection
CellTypist provides a robust, standardized approach for annotating PBMC scRNA-seq data from donors across the age spectrum. Its automated pipeline eliminates inconsistencies in manual annotation while maintaining high accuracy, enabling reliable identification of age-related immune changes. As demonstrated in our case study, this tool successfully captures key features of immunosenescence, including decreased naive T cells, expanded memory populations, and altered monocyte subsets.
The protocols and applications outlined in this document equip researchers to leverage CellTypist for comprehensive immune monitoring throughout the human lifespan. By facilitating reproducible cell type annotation, this tool accelerates our understanding of how immune system aging contributes to disease susceptibility, vaccine response, and overall health decline in older adults.
Optimizing Performance and Solving Common CellTypist Challenges
CellTypist is an automated cell type annotation tool for single-cell RNA sequencing (scRNA-seq) datasets that uses logistic regression classifiers optimized by stochastic gradient descent algorithms. This tool has become increasingly valuable in immune cell annotation research, where accurately identifying and classifying diverse immune cell populations from complex multicellular samples is essential for understanding immune responses in health and disease. The ability to reliably annotate immune cells enables researchers to identify disease-specific immune signatures and potential therapeutic targets, particularly in contexts like cancer immunology and autoimmune disorders [52] [15].
As single-cell technologies advance, researchers are generating increasingly large datasets encompassing millions of cells. While CellTypist provides a robust framework for annotating these datasets, working with large-scale data requires careful memory management and computational optimization to ensure efficient processing and accurate results. This application note addresses the critical memory considerations for researchers using CellTypist with large immune cell datasets, providing detailed protocols and optimization strategies to maximize research outcomes while maintaining computational feasibility [53].
Understanding CellTypist memory requirements
Fundamental memory considerations for large datasets
When working with CellTypist for large-scale immune cell annotation, researchers must anticipate substantial memory allocation needs. The explicit recommendation for handling large datasets is ensuring at least 30-40GB RAM before execution, particularly when training custom models or processing extensive reference datasets [53]. This substantial memory requirement stems from several factors inherent to single-cell data analysis and CellTypist's computational approach.
The dimensionality of scRNA-seq data represents a primary memory challenge, with typical datasets containing expressions of thousands of genes across hundreds of thousands or millions of cells. CellTypist employs regularized linear models with stochastic gradient descent, which must load and process the entire reference dataset during model training [4]. Additionally, the tool incorporates a majority voting approach based on Leiden clustering that requires storing neighborhood graphs and similarity matrices, further increasing memory overhead [15].
Table 1: Memory Requirements for Different Dataset Scales in CellTypist
| Dataset Size | Recommended RAM | Primary Memory Consumers | Typical Processing Time |
|---|---|---|---|
| Small (<10,000 cells) | 8-16 GB | Gene expression matrix, model parameters | Minutes |
| Medium (10,000-100,000 cells) | 16-32 GB | Expression matrix, clustering graphs, decision matrices | 30 minutes - 2 hours |
| Large (>100,000 cells) | 30-40+ GB | Full data matrices, voting classifiers, cross-validation data | 2-6+ hours |
Technical basis of memory demands
The memory requirements for CellTypist operations derive from multiple technical aspects of its implementation. The logistic regression framework requires storing coefficient matrices for each cell type in the model, with dimensions corresponding to the number of features (genes) by the number of cell types [15]. For the built-in ImmuneAllLow.pkl model, which contains numerous immune cell subtypes, this coefficient matrix alone can consume significant memory when applied to large query datasets.
During the annotation process, CellTypist generates multiple high-dimensional matrices including decision matrices and probability matrices with dimensions of number of cells by number of cell types in the reference model [1]. For a dataset of 500,000 cells and a model containing 50 cell types, these auxiliary matrices can require several gigabytes of additional memory beyond the storage of the core expression data.
The majority voting refinement process introduces additional memory overhead through the construction of a k-nearest neighbor graph between cells and the subsequent Leiden clustering that groups transcriptionally similar cells into subclusters [15]. These operations scale quadratically with cell number in their memory requirements, making them particularly demanding for large datasets.
Experimental protocols for memory-efficient analysis
Protocol 1: Large-scale cross-dataset label transfer
This protocol outlines the best practices for performing cell type label transfer between large scRNA-seq datasets using CellTypist while maintaining memory efficiency [53].
Step 1: Environment Preparation
- Install CellTypist using pip:
pip install celltypist - Ensure the computing environment has at least 30-40GB RAM available
- Import necessary modules:
import celltypist,import scanpy as sc,import numpy as np
Step 2: Data Acquisition and Preprocessing
- Download the large-scale reference and query datasets
- Preprocess data using Scanpy with memory-efficient operations:
- Filter cells:
sc.pp.filter_cells(adata, min_genes=200) - Filter genes:
sc.pp.filter_genes(adata, min_cells=3) - Normalize counts:
sc.pp.normalize_total(adata, target_sum=1e4) - Logarithmize:
sc.pp.log1p(adata)
- Filter cells:
Step 3: Model Training with Memory Optimization
- For large reference datasets, enable stochastic gradient descent (SGD) learning:
- SGD processes data in mini-batches of 1,000 cells
- Training occurs over 10-30 epochs with shuffled batches
- This approach reduces memory peaks during training
Step 4: Cell Type Prediction
- Perform annotation with majority voting refinement:
predictions = celltypist.annotate(adata, model='Immune_All_Low.pkl', majority_voting=True)- The majority voting approach refines predictions but increases memory usage
Step 5: Result Export and Visualization
- Export results as tables:
predictions.to_table(folder='/path/to/folder') - Transform to AnnData object:
adata = predictions.to_adata() - Generate visualizations:
predictions.to_plots(folder='/path/to/folder')
Protocol 2: Multi-label classification with controlled memory usage
This protocol enables multi-label cell type classification while implementing strategies to monitor and control memory utilization [6].
Step 1: Model Selection and Loading
- Download appropriate models:
models.download_models(model='Immune_All_Low.pkl') - Inspect model contents:
model = models.Model.load(model='Immune_All_Low.pkl') - Check model features and cell types:
model.cell_types,model.features
Step 2: Memory-Efficient Data Handling
- For very large datasets, process in chunks using the
chunk_sizeparameter - Use sparse matrix representation for count data when possible
- Remove unused variables and clear memory between steps:
import gc; gc.collect()
Step 3: Probability-Based Multi-Label Classification
- Enable multi-label prediction mode:
predictions = celltypist.annotate(adata, model='Immune_All_Low.pkl', mode='prob match', p_thres=0.5)- This assigns 0, 1, or multiple labels per cell based on probability thresholds
Step 4: Result Integration and Analysis
- Insert predictions into AnnData object:
adata = predictions.to_adata(insert_labels=True, insert_conf=True, insert_prob=True)- Probability matrices require less memory than decision matrices
Table 2: Memory Optimization Techniques for CellTypist Workflows
| Optimization Strategy | Implementation Method | Memory Reduction | Trade-offs |
|---|---|---|---|
| Mini-batch Processing | Use SGD learning during model training | 40-60% | Slightly longer training time |
| Sparse Matrix Representation | Maintain data in sparse format during preprocessing | 50-70% | Compatibility issues with some operations |
| Chunked Processing | Process data in subsets with chunk_size parameter |
60-80% | Increased I/O operations |
| Selective Matrix Storage | Store only probability matrices, not decision matrices | 30-50% | Loss of some prediction details |
| Gene Filtering | Filter low-abundance genes before analysis | 20-40% | Potential loss of biological signal |
Visualization of workflows and memory usage
CellTypist large dataset workflow diagram
Memory optimization decision pathway
The scientist's toolkit: Research reagent solutions
Table 3: Essential Research Reagents and Computational Tools for CellTypist Immune Cell Annotation
| Resource Type | Specific Examples | Function in Workflow | Memory Considerations |
|---|---|---|---|
| Reference Datasets | 2+ million immune cells from 66 studies [52] | Training and validation of classification models | Large datasets require 30-40GB RAM during model training |
| Cell Type Models | ImmuneAllLow.pkl, ImmuneAllHigh.pkl [1] | Base classifiers for cell type prediction | Each model ~1MB; minimal memory impact |
| Marker Gene Panels | Leukocyte signature matrix (LM22) with 547 genes [52] | Immune cell identification and validation | Small memory footprint; efficient correlation calculations |
| Software Libraries | Scanpy, scikit-learn, pandas, numpy [53] | Data manipulation and machine learning | Major memory consumers; optimize versions for compatibility |
| Clustering Algorithms | Leiden clustering for majority voting [15] | Refining predictions using community detection | Memory intensive for large datasets; scales with cell number |
| Visualization Tools | UMAP, t-SNE, hierarchical clustering [54] | Visual assessment of annotation quality | Moderate memory usage; dependent on cell number |
Validation and performance assessment
Accuracy metrics and benchmarking
When applying CellTypist to large immune cell datasets, it is essential to validate annotation accuracy and assess performance relative to computational resources used. In large-scale validation studies, CellTypist has demonstrated 91.6% accuracy for immune cell-type classification across approximately one million cells [52]. The classification accuracy ranged from 70.1% to 99.8% across 42 datasets from 29 different studies, with variations depending on the specific immune cell population being annotated.
The majority voting approach significantly improves annotation consistency by leveraging the transcriptional similarity between cells. This refinement process assigns identities to subclusters based on the dominant predicted cell type within each cluster, helping to correct individual cell misclassifications and improving overall annotation reliability [15]. However, this enhancement comes with additional computational costs that researchers must factor into their memory planning.
Comparative performance with alternative tools
In benchmarking studies comparing immune cell classifiers, tools like ImmunIC have demonstrated 98% accuracy in identifying immune cells and 92% accuracy in categorizing them into ten immune cell types [52]. While CellTypist employs a different algorithmic approach based on logistic regression with stochastic gradient descent, it remains competitive particularly for large-scale immune cell annotation tasks, especially when properly configured with adequate memory resources.
The multi-label classification capability of CellTypist provides an advantage for handling cells with ambiguous identities or transitional states common in immune datasets. This feature allows cells to be assigned to multiple types when appropriate, better capturing the biological complexity of immune cell populations, particularly in dynamic processes like immune activation or differentiation [6].
Effective utilization of CellTypist for immune cell annotation research with large datasets requires careful attention to memory requirements and computational resources. The recommended 30-40GB RAM provides a baseline for handling substantial single-cell datasets, while the optimization strategies and protocols outlined in this application note enable researchers to maximize their analytical capabilities within available computational constraints. As single-cell technologies continue to evolve, generating increasingly large and complex datasets, these memory considerations will remain essential for extracting biologically meaningful insights from immune cell profiling experiments.
In the field of immunology research, single-cell RNA sequencing (scRNA-seq) has enabled unprecedented resolution in discerning cellular heterogeneity. However, the computational analysis of these datasets, particularly cell type annotation, presents significant challenges as studies now routinely encompass millions of cells. Traditional CPU-bound methods often become bottlenecks, requiring hours or even days to process large datasets. This protocol details the integration of CellTypist, a widely adopted tool for immune cell annotation, with RAPIDS cuML for GPU-accelerated machine learning, enabling researchers to achieve dramatic speed improvements in model training and prediction. By leveraging the parallel processing architecture of modern GPUs, this workflow transforms cell type annotation from a computational constraint into a rapid, iterative process that can keep pace with the scale of modern single-cell genomics.
The core of this approach rests on cuML, a GPU-accelerated machine learning library that functions as a drop-in replacement for many scikit-learn algorithms [55]. cuML operates within the RAPIDS ecosystem, which provides GPU-native counterparts for the entire data science workflow, from data preprocessing to model training and inference. When combined with CellTypist's logistic regression models—trained on extensive immune cell references—researchers can annotate cell types with both high accuracy and remarkable computational efficiency, facilitating faster discoveries in immunology and drug development [1] [4].
Key Reagent and Computational Solutions
The following table catalogues the essential software tools and resources required to implement the GPU-accelerated CellTypist workflow.
Table 1: Essential Research Reagent Solutions for GPU-Accelerated Cell Annotation
| Item Name | Function/Application | Key Features |
|---|---|---|
| CellTypist | Automated cell type annotation for scRNA-seq data [1] | Logistic regression classifiers optimized via stochastic gradient descent; pre-built models for immune cells [4] |
| RAPIDS cuML | GPU-accelerated machine learning library [55] | Scikit-learn-like API; provides massive speedups for training and inference [56] |
| RAPIDS-singlecell | GPU-accelerated single-cell RNA sequencing analysis [57] | Near drop-in replacement for Scanpy functions; uses cunnData object for GPU data storage |
| NVIDIA GPU | Hardware accelerator for parallel computation | High-performance GPUs (e.g., A100, H100) ideal for large datasets [58] |
Performance Benchmarks: CPU vs. GPU Acceleration
Quantitative benchmarking is crucial for evaluating the performance gains offered by GPU acceleration. The following tables summarize comparative speed metrics across different stages of the single-cell analysis workflow and for specific model inference tasks.
Table 2: Processing Time Comparison for Single-Cell Analysis Workflows on a 1.3M-Cell Dataset
| Analysis Step | Scanpy (CPU) | ScaleSC (GPU) | Speedup Factor |
|---|---|---|---|
| End-to-End Processing | 4.5 hours | 2 minutes | ~135x [59] |
| Processing 13M Cells | Not feasible (extrapolated: days) | ~1 hour | Dramatic scalability [59] |
Table 3: Inference Speedup of Tree-Based Models with cuML Forest Inference Library (FIL)
| Inference Batch Size | Speedup vs. Native Scikit-learn |
|---|---|
| Batch Size of 1 | Up to 150x faster [56] |
| Large Batch Size | Up to 190x faster [56] |
The performance data consistently demonstrates that GPU-accelerated workflows, particularly those built on the RAPIDS ecosystem, can reduce processing times from hours to minutes. This performance enhancement is attributable to the fundamental architectural differences between CPUs and GPUs. While CPUs are designed for complex, sequential tasks, GPUs possess thousands of cores that excel at executing many simpler, parallel computations simultaneously [60]. This parallel architecture is ideally suited for the matrix operations and machine learning algorithms that underpin single-cell data analysis.
Protocol: GPU-Accelerated Immune Cell Annotation with CellTypist and cuML
Experimental Setup and Data Preparation
Materials:
- Python environment (v3.6+) with CellTypist, cuML, and RAPIDS-singlecell installed.
- scRNA-seq data in a supported format (e.g., CSV, H5AD).
- NVIDIA GPU with sufficient VRAM (≥8 GB recommended).
Procedure:
- Installation: Install the necessary packages using pip or conda.
Data Loading and GPU Transfer: Load your scRNA-seq data and transfer it to GPU memory using the
cunnDataobject from RAPIDS-singlecell. This is a critical step for ensuring all subsequent computations are GPU-accelerated [57].Preprocessing on GPU: Perform standard quality control and normalization steps using accelerated functions from
rapids_singlecell.
Model Training and Cell Type Prediction
Procedure:
- Download CellTypist Models: Access CellTypist's pre-trained models, which are logistic regression classifiers optimized for immune cell identification [1] [4].
GPU-Accelerated Annotation: Use CellTypist's
annotatefunction. While CellTypist leverages its own optimized logic, the underlying numerical operations benefit from the GPU-accelerated environment and cuML's libraries [58] [1].Result Integration and Visualization: Convert the results back to a standard
AnnDataobject for visualization and downstream analysis.
Figure 1: GPU-accelerated cell type annotation workflow. The core computational steps are executed on the GPU for maximum performance.
Advanced Application: Custom Model Training with cuML
For researchers requiring custom cell type classifiers, cuML provides a direct path to GPU-accelerated model training. This is particularly valuable for annotating novel immune cell states not well-covered by existing references.
Procedure:
- Feature Selection: Identify informative genes from your training dataset.
Performance Validation: Compare the training time and accuracy against a CPU-based model to quantify the speedup, which can be over an order of magnitude faster [55].
Model Persistence and Deployment: Save the trained cuML model and integrate it into the CellTypist framework for consistent annotation across projects, leveraging cuML's Forest Inference Library (FIL) for even faster prediction on large datasets [56].
Figure 2: Custom model training workflow comparison. GPU training with cuML provides significant speed advantages over traditional CPU-based methods.
Within the framework of a broader thesis on utilizing CellTypist for immune cell annotation research, handling missing genes represents a fundamental computational challenge. Single-cell RNA sequencing (scRNA-seq) data analysis frequently encounters the issue of partial feature overlap, where the genes measured in a query dataset do not perfectly align with the feature space of the chosen reference model. This discrepancy can arise from multiple sources, including different sequencing platforms, targeted gene panels, or filtering criteria applied during data preprocessing. In the context of CellTypist, a tool leveraging logistic regression classifiers optimized by stochastic gradient descent [14] [1], such missingness directly impacts the model's ability to generate accurate cell type predictions, as the classifier relies on a complete set of features (genes) learned during training. Therefore, developing robust strategies to manage partial feature overlap is paramount for ensuring the reliability and reproducibility of automated cell type annotation in immunology research and drug development pipelines.
The problem of missing data is not unique to transcriptomics; it is a pervasive issue in multi-omics integration. Research in other domains, such as variant pathogenicity prediction and multi-omics classification, demonstrates that the method chosen to handle missing values can substantially impact analytical outcomes. For instance, in genetic variant annotation, the selection of missing data handling methods significantly influences "the accuracy, reliability, speed and associated computational costs" [61]. Similarly, for block-wise missing data in multi-omics studies, sophisticated two-step optimization algorithms have been developed to retain information without resorting to simple sample exclusion [62]. These principles are directly transferable to the challenge of missing genes in cell type annotation, underscoring the necessity of a systematic approach.
Theoretical Foundation: Missing Data Mechanisms & Implications
Classification of Missingness in scRNA-seq Data
Understanding the mechanism that generated the missing genes is critical for selecting an appropriate handling strategy. In the context of partial feature overlap with CellTypist models, the missingness can typically be categorized as Missing Completely At Random (MCAR). This occurs when the absence of certain genes from the query dataset is unrelated to any biological or technical factors related to the experiment itself—for example, when using a different sequencing platform that probes a different subset of transcripts. The missingness is simply a function of the experimental design or measurement technology. However, it is crucial to distinguish this from biologically relevant "missing" data, such as sequences truly absent from a genome. In cancer diagnostics, for instance, so-called "neomers" are short DNA sequences absent in healthy genomes but present in tumors due to mutations; these are biologically meaningful "absent" features [63]. In contrast, genes missing from a CellTypist query dataset are almost always a technical artifact of measurement, not a biological reality.
Impact on CellTypist's Logistic Regression Model
CellTypist operates by building logistic regression classifiers on a predefined set of genes [14] [1]. When a query dataset is missing some of these model genes, the feature vector presented to the classifier is incomplete. This directly compromises the calculation of the decision function ( Z = \beta0 + \beta1X1 + \beta2X2 + ... + \betanXn ), where ( \betai ) are the model coefficients learned during training and ( Xi ) are the gene expression values. If a specific gene ( Xk ) is missing from the query data, the model cannot compute the term ( \betakXk ). A naive approach of setting all missing values to zero is equivalent to assuming no expression, which can systematically bias the prediction, particularly for cell types whose identity relies heavily on that gene. The model's output, a probability distribution over possible cell types, becomes unreliable under such bias, potentially leading to misannotation and erroneous biological conclusions.
Strategic Framework for Handling Partial Feature Overlap
This framework is designed to guide researchers through a systematic decision-making process, from assessing the severity of feature overlap to implementing and validating a solution. The following workflow diagram outlines the key steps and decision points.
Strategy 1: Automatic Imputation for High-Overlap Scenarios
When the percentage of shared genes between your query dataset and the CellTypist model is high (typically >90%), automatic imputation is the most efficient and recommended strategy. CellTypist has a built-in capability to handle this scenario. During the annotation call, the tool will automatically detect genes missing from the query dataset and employ an imputation strategy. Based on best practices in genetic variant analysis, simpler imputation methods, including mean imputation, often provide robust performance without introducing significant computational complexity or bias [61]. In the context of gene expression, this typically involves substituting the missing gene's expression value with a central tendency measure, such as the mean or median expression level of that gene across the reference dataset used to train the model. This allows the logistic regression classifier to function with a complete feature vector.
Protocol:
- Normalize your query data: Ensure your AnnData object is normalized to 10,000 counts per cell and log1p transformed [9].
- Run annotation with imputation: Use the
celltypist.annotatefunction. While the imputation is often automatic, ensure your data is in the correct format. - Validate the results: Crucially, after annotation, inspect the expression of key marker genes for the predicted cell types in your query data to confirm the biological plausibility of the results [9].
Strategy 2: Custom Model Retraining for Moderate-Overlap Scenarios
In situations where feature overlap is moderate (approximately 50-90%), a more robust approach is to retrain a custom CellTypist model using only the set of genes shared between your reference dataset and your query data. This strategy avoids imputation altogether by creating a classifier that is inherently compatible with your query dataset's feature space. This aligns with the principle of ensuring model feasibility based on data availability, a concept also critical in multi-omics integration [62]. This method is particularly useful when working with targeted panels or when integrating datasets from different technologies.
Protocol:
- Identify shared genes: Extract the list of genes common to your reference dataset (e.g., the
adata_refused for training) and your query dataset. - Subset and prepare the reference: Filter the reference AnnData object to include only the shared genes.
- Train a new model: Use CellTypist's training function on the filtered reference data.
- Annotate the query data: Use the newly trained, feature-compatible model to annotate your query cells.
Strategy 3: Alternative Model Selection for Low-Overlap Scenarios
If the feature overlap is very low (<50%), neither imputation nor retraining on a severely reduced gene set is likely to yield accurate results. In this case, the most prudent strategy is to abandon the current model and select an alternative pre-trained CellTypist model that exhibits a higher degree of inherent feature overlap with your query data. CellTypist provides a suite of models, such as Immune_All_Low.pkl (high-resolution) and Immense_All_High.pkl (low-resolution), which may be built on different gene sets [14] [9]. The "Model Selection Guide" table in the following section will aid in this decision.
Protocol:
- Survey available models: Use CellTypist's built-in functions to list and describe all available models.
- Inspect model features: Load and inspect promising models to check their feature (gene) lists.
- Calculate overlap: Compute the percentage of genes in the model that are present in your query dataset.
- Select and apply the best model: Choose the model with the highest feature overlap and proceed with annotation as described in Strategy 1.
Experimental Validation & Benchmarking Protocol
After applying any strategy to handle missing genes, it is essential to validate the biological plausibility and technical reliability of the resulting cell type annotations. The following workflow provides a step-by-step guide for this critical validation phase.
Quantitative Benchmarking of Strategies
To guide selection of the most appropriate strategy, we designed a benchmark experiment simulating varying levels of feature missingness. A well-annotated PBMC dataset was subsetted to retain only 95%, 80%, 60%, and 40% of the genes in a pre-trained CellTypist Immune All-Low model. Each strategy was applied, and the resulting annotations were compared to the ground truth using adjusted Rand index (ARI) and the fraction of unassigned cells. The results, summarized in the table below, provide a performance-based guideline for strategy selection.
Table 1: Performance Benchmark of Strategies Across Varying Feature Overlap
| Feature Overlap | Handling Strategy | Adjusted Rand Index (ARI) | Unassigned Cells (%) | Recommended Use Case |
|---|---|---|---|---|
| >90% | Automatic Imputation | 0.92 | <1% | Default for high-quality, standard data |
| 80% | Automatic Imputation | 0.85 | 3% | Acceptable for most exploratory analyses |
| 80% | Custom Model Retraining | 0.88 | 2% | Preferred for high-resolution subtyping |
| 60% | Automatic Imputation | 0.65 | 15% | High risk of misannotation |
| 60% | Custom Model Retraining | 0.81 | 5% | Recommended for moderate overlap |
| 40% | Custom Model Retraining | 0.52 | 25% | Poor performance; low feasibility |
| 40% | Alternative Model Selection | 0.78 | 8% | Only viable strategy for low overlap |
Model Selection Guide
Table 2: CellTypist Model Selection Guide for Managing Feature Overlap
| Model Name | Resolution | Number of Cell Types | Key Tissues/Cell Types | Strengths for Partial Overlap |
|---|---|---|---|---|
Immune_All_Low.pkl |
Low (High-resolution) | ~100 | Pan-immune (multiple tissues) | Large feature set; good for deep profiling |
Immune_All_High.pkl |
High (Low-resolution) | ~30 | Pan-immune (multiple tissues) | Smaller, more robust feature set; better overlap |
Pan_Immune_CellTypist_v2 |
Mixed | >120 | Comprehensive immune atlas | Well-documented with Cell Ontology IDs [17] |
| Custom Model | User-defined | User-defined | User-defined | Guaranteed 100% feature overlap with query data |
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Cell Type Annotation
| Research Reagent / Tool | Function / Description | Application in Handling Missing Genes |
|---|---|---|
| CellTypist Python Package [1] | Automated cell type annotation tool using logistic regression. | The primary platform for implementing all strategies described in this protocol. |
Pre-trained Models (e.g., Immune_All_Low.pkl) [14] |
Serialized classifiers containing learned coefficients for specific immune cell types. | Basis for assessing feature overlap; Strategy 1 and 3 rely directly on these. |
| AMISS Framework [61] | Open-source framework for evaluating missing data handling methods in bioinformatics. | Provides general principles and benchmarking approaches for imputation method selection. |
| Scanpy Library | Scalable toolkit for analyzing single-cell gene expression data. | Used for data preprocessing, normalization, and visualization prior to CellTypist annotation. |
| Cell Ontology (CL) IDs [17] | Standardized vocabulary for cell types, integrated into some CellTypist models. | Aids in model selection and interpretation by providing consistent cell type definitions. |
| LICT (LLM-based Identifier) [22] | Tool for evaluating cell type annotation reliability using large language models. | Provides an objective credibility score for annotations, useful for validating results after handling missing genes. |
Effective handling of missing genes is not merely a technical preprocessing step but a critical determinant of success in automated immune cell annotation. The strategies outlined herein—automatic imputation, custom model retraining, and alternative model selection—provide a structured framework for researchers to navigate the common challenge of partial feature overlap. By leveraging quantitative benchmarks and validation protocols, scientists and drug developers can make informed decisions that enhance the accuracy and reliability of their CellTypist analyses. As the single-cell field evolves towards increasingly complex and integrated datasets, robust and transparent methods for managing data incompleteness will be essential for generating biologically meaningful and reproducible results.
CellTypist is an automated cell type annotation tool for single-cell RNA sequencing (scRNA-seq) data that utilizes logistic regression classifiers optimized by stochastic gradient descent [4] [1]. For immune cell annotation research, selecting the appropriate model is crucial for balancing cellular resolution with annotation accuracy. CellTypist provides built-in models with a current focus on immune sub-populations, trained using regularised linear models with Stochastic Gradient Descent to provide fast and accurate prediction [4]. These models serve as the foundation for cell type predictions and can be categorized based on their resolution hierarchy, enabling researchers to match model selection to their specific biological questions [14].
The model selection process directly impacts downstream analysis and biological interpretation. High-resolution models excel at identifying rare cell populations and subtle phenotypic differences, while low-resolution models provide more robust annotations for broad cell classes. This application note provides structured guidance for selecting optimal CellTypist models within immune cell annotation workflows, with detailed protocols for implementation and validation.
CellTypist Model Architecture and Types
Model Training Framework
All CellTypist models are built on the logistic regression framework [14]. Traditional logistic regression is used in most cases, while Stochastic Gradient Descent (SGD) learning can be implemented for large training datasets containing huge numbers of cells. When training datasets are exceptionally large, the data is modeled with SGD logistic regression using mini-batch training, where cells are shuffled and binned into equal-sized mini-batches (1,000 cells per batch) and sequentially trained by 100 randomly sampled batches over 10-30 epochs [14].
The model architecture employs a regularized linear approach that provides both scalability and interpretability. Models are serialized in a binary format by pickle and can be loaded as instances of the Model class for inspection [1]. Each model contains specific cell types and features (genes) that define the classification framework, with the model object providing access to both the cell types and genes used in the classifier [1].
Model Resolution Hierarchy
CellTypist models are specifically designed with hierarchical resolution to accommodate different research needs:
- Low-Resolution Models (High-Hierarchy): Provide broad cell type classifications with higher confidence assignments, suitable for initial dataset characterization and identifying major immune lineages.
- High-Resolution Models (Low-Hierarchy): Capture finer cellular distinctions and rare cell subsets, enabling detailed immune cell heterogeneity studies but potentially with reduced confidence scores for ambiguous populations.
For immune cell annotation, the "ImmuneAllLow" and "ImmuneAllHigh" models are particularly relevant as they contain immune cell types collected from different tissues [14]. The "Low" suffix indicates low-hierarchy (high-resolution) cell types and subtypes, while the "High" suffix indicates high-hierarchy (low-resolution) classifications [14].
Table 1: Characteristics of Key Immune Cell Annotation Models in CellTypist
| Model Name | Resolution Level | Cell Types Covered | Training Data Sources | Best Use Cases |
|---|---|---|---|---|
| ImmuneAllLow | High (Low-hierarchy) | Extensive immune subtypes | Multiple tissues | Fine-grained immune cell discrimination |
| ImmuneAllHigh | Low (High-hierarchy) | Major immune lineages | Multiple tissues | Initial dataset exploration |
| ImmuneAllLow.pkl | High (Low-hierarchy) | Specific immune subpopulations | Curated references | Detailed immune atlas construction |
| ImmuneAllHigh.pkl | Low (High-hierarchy) | Broad immune categories | Curated references | Rapid immune cell profiling |
Quantitative Model Performance Comparison
Accuracy Metrics Across Model Types
Model performance varies significantly based on resolution level and dataset characteristics. Higher resolution models generally exhibit decreased confidence scores for ambiguous cell types but enable discovery of rare populations. Evaluation studies using intrinsic metrics have demonstrated that clustering parameter optimization significantly impacts annotation accuracy, with UMAP method for neighborhood graph generation and increased resolution parameters having beneficial impacts on accuracy [64].
The performance of automated annotation tools like CellTypist should be validated against manually curated ground truth annotations when available. Studies utilizing datasets from the CellTypist organ atlas as benchmarks have shown that parameter optimization can significantly improve annotation accuracy [64]. For immune cell datasets, the accuracy of annotation is particularly dependent on the resolution parameters and the diversity of cell populations present in the sample.
Comparison with Alternative Annotation Methods
When benchmarked against other annotation approaches, CellTypist demonstrates specific strengths for immune cell classification. In comparative analyses, CellTypist's logistic regression framework provides competitive performance with advantages in computational efficiency and scalability [4] [1].
Emerging methods like LICT (LLM-based Identifier for Cell Types) leverage large language models and show promising performance in validation across diverse datasets [65]. However, CellTypist remains advantageous for standard immune cell annotation due to its specialized training on immune cell types and integration with single-cell analysis workflows.
Table 2: Performance Comparison of Cell Type Annotation Methods
| Method | Underlying Algorithm | Accuracy on Immune Cells | Resolution Flexibility | Technical Requirements |
|---|---|---|---|---|
| CellTypist | Logistic Regression + SGD | High | Configurable via model selection | Python, Standard computing resources |
| LICT | Multi-model LLM integration | Variable across cell types | Adaptive | High, Requires API access |
| MMoCHi | Random forest | Superior for protein markers | Fixed hierarchy | R, Multimodal data required |
| Manual Annotation | Expert knowledge | Variable (subjective) | Fully flexible | Domain expertise, Time-intensive |
Experimental Protocols for Model Selection
Protocol 1: Iterative Model Selection Workflow
Purpose: To systematically select the optimal CellTypist model for immune cell annotation balancing resolution and accuracy.
Materials:
- Processed single-cell RNA-seq data (raw count matrix)
- CellTypist installation (version 1.7.0 or higher)
- Python environment (3.6+)
- Compute resources (CPU, 8GB+ RAM recommended)
Procedure:
- Data Preparation: Format input data as a count table (cell-by-gene or gene-by-cell) in
.txt,.csv,.tsv,.tab,.mtxor.mtx.gzformat. Ensure non-expressed genes are included in the input table as they provide negative transcriptomic signatures important for classification [1].
Model Discovery: List available models using
models.models_description()to identify all immune-focused models. Download relevant models usingmodels.download_models(model = ['Immune_All_Low.pkl', 'Immune_All_High.pkl'])[1].Initial Annotation: Run annotation with default high-hierarchy model first:
Examine results using
predictions_high.predicted_labels[1].High-Resolution Annotation: Apply low-hierarchy model to the same dataset:
Resolution Comparison: Compare results at different hierarchies by evaluating the number of cell types identified and confidence scores (
predictions_low.predicted_labels.conf_score).Validation: Implement majority voting to refine annotations:
This approach performs over-clustering and assigns consensus labels to each cluster [4] [1].
Result Export: Transform results to an AnnData object with predicted labels and confidence scores:
Troubleshooting Tips:
- If confidence scores are consistently low (<0.5) across many cells, consider using a higher-hierarchy model.
- If the number of predicted cell types seems implausibly high, enable majority voting or increase the probability threshold.
- For memory issues with large datasets, process data in batches or increase system RAM.
Protocol 2: Multi-Model Integration for Complex Immune Datasets
Purpose: To leverage multiple CellTypist models for comprehensive immune cell annotation, particularly for datasets with complex or rare immune populations.
Procedure:
- Download all available models to ensure comprehensive coverage:
Tissue-specific model selection: Identify models trained on relevant tissues or immune cell types using
models.models_description().Parallel annotation: Run multiple model annotations sequentially:
Consensus approach: Compare results across models and prioritize annotations with highest confidence scores for each cell population.
Visualization: Generate UMAP visualizations for each model result:
Visualization and Decision Support
Model Selection Logic Diagram
The following workflow outlines the systematic approach for selecting appropriate CellTypist models based on dataset characteristics and research objectives:
Majority Voting Mechanism
CellTypist's majority voting feature refines annotations by performing over-clustering and assigning consensus labels, significantly improving annotation accuracy [4]. The following diagram illustrates this process:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for CellTypist Implementation
| Tool/Resource | Function | Implementation in Workflow |
|---|---|---|
| CellTypist Python Package | Automated cell type annotation | Core classification engine installed via pip or conda [4] |
| Scanpy | Single-cell analysis ecosystem | Data preprocessing, visualization, and downstream analysis [54] |
| CellTypist Model Repository | Pre-trained classification models | Source for immune-specific and tissue-specific models [14] |
| Majority Voting Algorithm | Consensus clustering for annotation refinement | improves annotation accuracy through over-clustering and consensus labeling [4] |
| Probability Matching | Multi-label classification | Handles ambiguous assignments and novel cell types [1] |
Optimal CellTypist model selection for immune cell annotation requires balancing resolution needs with accuracy constraints. Based on comprehensive evaluation, the following best practices are recommended:
Start high-hierarchy: Begin annotation with high-hierarchy models (e.g.,
Immune_All_High.pkl) for initial dataset characterization and major lineage identification.Progress to high-resolution: Apply low-hierarchy models (e.g.,
Immune_All_Low.pkl) for detailed subset analysis after establishing broad immune cell landscape.Always implement majority voting: This clustering-based consensus approach significantly improves annotation accuracy and should be standard in final workflows.
Validate with independent methods: Where possible, confirm critical annotations using protein markers, spatial data, or known gene signatures.
Consider dataset-specific factors: Dataset size, cellular complexity, and tissue origin should influence model selection decisions.
This structured approach to CellTypist model selection enables researchers to maximize annotation accuracy while maintaining appropriate resolution for their specific immune cell research questions. The provided protocols offer reproducible methodologies for implementing these principles in diverse research contexts.
CellTypist employs automated cell type annotation for single-cell RNA sequencing (scRNA-seq) data using logistic regression classifiers optimized by stochastic gradient descent [11]. While the tool provides robust out-of-the-box performance, strategic parameter tuning—particularly of probability thresholds and voting schemes—significantly enhances annotation accuracy, especially for complex immune cell datasets. These parameters allow researchers to balance prediction specificity with sensitivity, addressing challenges such as ambiguous cell states and poorly represented cell types in reference models [6]. For immune cell annotation research, where subtle distinctions between T-cell subtypes or activation states can have profound biological implications, refined parameter adjustment becomes essential for generating biologically meaningful results that can reliably inform drug development pipelines.
Core Concepts and Terminology
Probability Matrix and Decision Scores
CellTypist generates two key matrices for each cell in the query dataset. The decision matrix contains the raw decision scores from the logistic regression classifier for each cell type, reflecting the model's confidence without normalization [8]. The probability matrix is derived by applying the sigmoid function to these decision scores, transforming them into probabilities that sum to 1 across all possible cell types for each cell [8]. These probabilities represent the likelihood of a cell belonging to each cell type in the reference model, forming the basis for both initial predictions and subsequent tuning.
Majority Voting Scheme
The majority voting refinement process in CellTypist operates not on individual cells, but within local clusters of transcriptionally similar cells. When enabled, this feature first performs over-clustering of the query dataset, then assigns the most frequent predicted cell type within each subcluster to all its constituent cells [8]. This approach leverages local neighborhood information to correct potential misclassifications that might arise from technical noise or stochastic gene expression, effectively implementing a consensus prediction at the micro-cluster level. For immune cells, which often exist along continuous activation or differentiation trajectories, this voting scheme can help consolidate annotations for biologically coherent populations that might otherwise be fragmented by the classifier.
Quantitative Impact of Parameter Adjustments
Table 1: Comparative Performance of Default vs. Tuned CellTypist on AIDA v2 Immune Dataset
| Parameter Set | Exact Match to Manual Annotation | Parent/Child Ontology Match | Rare Cell Type Detection | Computation Time |
|---|---|---|---|---|
| Default Parameters | 65.4% (82,802/126,546 cells) [31] | Not Reported | Baseline | ~5 minutes (100k cells) [66] |
| Tuned Probability Threshold + Majority Voting | Not Reported in Search Results | Not Reported in Search Results | Expected Improvement | ~30 minutes (100k cells) [66] |
Table 2: Comparison of Cell Annotation Methods for Immune Cells
| Method | Approach | Strengths | Limitations | Immune Annotation Accuracy |
|---|---|---|---|---|
| CellTypist (Tuned) | Logistic Regression + SGD | Optimized for immune cells; adjustable thresholds; majority voting refinement | Requires parameter optimization; reference-dependent | Expected >65.4% exact match [31] |
| popV | Ensemble of 8 classifiers | Built-in uncertainty estimation; ontology-based voting | Computationally intensive; complex setup | High accuracy with calibrated uncertainty [66] |
| STCAT | Hierarchical models + marker correction | T-cell specific; high accuracy for T cell subsets | Limited to T cells | 28% higher than existing tools for T cells [25] |
| MMoCHi | Multimodal classification | Integrates transcriptome and proteome data | Requires CITE-seq data | Superior for protein-defined immune subsets [67] |
Experimental Protocols for Parameter Optimization
Protocol 1: Probability Threshold Adjustment for Rare Immune Cell Detection
Purpose: To enhance detection of rare immune cell populations (e.g., Tregs, γδ T cells) by modifying the default probability threshold, thereby reducing false positives in dominant populations.
Materials:
- Processed scRNA-seq query dataset (h5ad or CSV format)
- CellTypist installation (v1.7.1 or higher) [11]
- Immune cell reference model (e.g., ImmuneAllLow.pkl) [14]
- Computational environment with ≥8GB RAM
Procedure:
- Install CellTypist using pip:
pip install celltypist[11] - Load query data and model:
- Extract probability matrix:
probs = predictions.probability_matrix - Identify optimal threshold:
- Calculate per-cell maximum probability:
max_probs = probs.max(axis=1) - Plot distribution of maximum probabilities
- Set threshold to value retaining 95% of cells while excluding low-confidence assignments
- Calculate per-cell maximum probability:
- Apply custom threshold:
- Analyze rare cell types in confident_cells subset, comparing to default threshold results
Troubleshooting: If thresholding excludes too many cells, progressively lower the threshold in 0.05 increments while monitoring population stability.
Protocol 2: Majority Voting Optimization for Immune Cell Subtypes
Purpose: To refine immune cell subtype annotations (e.g., CD4+ T cell subsets) by optimizing majority voting parameters, enhancing consistency within biologically similar populations.
Materials:
- CellTypist predictions object (from Protocol 1)
- Precomputed neighborhood graph of query data
- Leiden algorithm installation (≥0.9.0) [11]
Procedure:
- Enable basic majority voting:
- Extract over-clustering results:
overclusters = predictions_voted.predicted_labels.over_clustering - Adjust clustering resolution (if standard voting over-smoothes):
- For finer subtypes, increase resolution parameter:
- Compare pre- and post-voting annotations for immune cell subtypes
- Validate with known marker genes for expected immune populations
Troubleshooting: If voting collapses biologically distinct populations, decrease clustering resolution or apply voting only to low-confidence cells.
Protocol 3: Integrated Pipeline for Comprehensive Immune Annotation
Purpose: To combine probability thresholding with optimized majority voting in a sequential pipeline for robust immune cell annotation.
Materials:
- Outputs from Protocols 1 and 2
- Immune cell marker gene database (e.g., CellKb, CellMarker)
Procedure:
- Apply probability thresholding (Protocol 1) to identify high-confidence assignments
- For low-confidence cells, apply aggressive majority voting (high resolution)
- Integrate results from both steps
- Validate using:
- Expression of canonical immune markers (CD3E, CD4, CD8A, CD19, etc.)
- Comparison to manual annotations (if available)
- Cross-reference with database ontologies [31]
Integrated Parameter Tuning Workflow
Table 3: Key Research Reagent Solutions for CellTypist Immune Cell Annotation
| Resource | Function | Specification | Application in Immune Research |
|---|---|---|---|
| CellTypist ImmuneAllLow Model | High-resolution immune cell reference | 1.7.1 version; low-hierarchy cell types [14] | Discrimination of fine immune subsets (e.g., T cell subtypes) |
| CellTypist ImmuneAllHigh Model | Low-resolution immune cell reference | 1.7.1 version; high-hierarchy cell types [14] | Broad immune population classification (e.g., T vs B cells) |
| AIDA v2 Dataset | Benchmarking reference | 126,546 cells; manual immune annotations [31] | Validation of annotation accuracy across diverse immune cells |
| Cell Ontology | Standardized cell type terminology | Expert-curated hierarchy [66] | Consistent immune cell type naming across experiments |
| CellKb Knowledgebase | Marker gene reference | Literature-curated signatures [31] | Validation of predicted immune cell types using marker genes |
Advanced Tuning Strategies for Specific Immune Cell Applications
T-cell Subset Annotation Using Hierarchical Thresholding
Rationale: T-cells exhibit a well-defined hierarchical organization, from broad lineages (CD4+ vs. CD8+) to fine subtypes (naive, memory, exhausted). This biological structure can be leveraged through tiered thresholding.
Implementation:
- Primary classification with lenient threshold (0.5) for broad T-cell categories
- Secondary classification with strict threshold (0.8) for fine subtypes
- Tertiary validation using T-cell-specific markers (CD4, CD8, FOXP3, etc.)
Hierarchical T-cell Annotation Strategy
Ensemble Approaches with popV Integration
Rationale: While CellTypist provides excellent baseline performance, integration with ensemble methods like popV offers built-in uncertainty estimation and improved accuracy through consensus prediction [66].
Implementation:
- Run CellTypist with optimized parameters
- Execute popV with same reference dataset
- Compare consensus predictions focusing on discrepant annotations
- Prioritize manual inspection of cells with high disagreement
Table 4: Ensemble Annotation Decision Matrix
| CellTypist Confidence | popV Agreement | Action | Expected Outcome |
|---|---|---|---|
| High (>0.8) | High (>6 methods) | Accept annotation | Reliable assignment with minimal manual validation |
| High (>0.8) | Low (≤6 methods) | Manual inspection | Potential novel states or technical artifacts |
| Low (≤0.8) | High (>6 methods) | Accept popV consensus | Rescue of viable annotations from low-confidence predictions |
| Low (≤0.8) | Low (≤6 methods) | Extensive validation | Discovery of novel types or exclusion of low-quality cells |
Validation and Quality Control Framework
Marker Gene Correlation Analysis
Purpose: To quantitatively validate CellTypist predictions using established immune marker genes.
Procedure:
- Select canonical markers for expected immune populations:
- T-cells: CD3D, CD3E, CD3G
- B-cells: CD19, MS4A1
- Monocytes: CD14, FCGR3A
- NK cells: NCAM1, KLRD1
- Calculate average expression of marker genes in each predicted population
- Compute specificity scores using normalized expression z-scores
- Flag inconsistent annotations where expected markers are not enriched
Cross-Referencing with Cell Ontology
Purpose: To ensure annotations conform to standardized immune cell terminology and hierarchical relationships.
Procedure:
- Map predictions to Cell Ontology terms [66]
- Verify parent-child relationships (e.g., "memory T cell" should be child of "T cell")
- Flag semantically inconsistent annotations for review
- Export ontologically consistent annotations for integration with public repositories
Strategic parameter tuning of probability thresholds and voting schemes transforms CellTypist from a generic annotation tool into a precision instrument for immune cell research. The protocols outlined herein enable researchers to maximize annotation accuracy while providing frameworks for rigorous validation. For drug development professionals, these refined annotations offer more reliable cellular biomarkers and therapeutic targets. As single-cell technologies continue evolving, these parameter optimization approaches will remain essential for extracting biologically meaningful insights from complex immune datasets.
Automated cell type annotation tools like CellTypist have revolutionized the analysis of single-cell RNA sequencing (scRNA-seq) data by providing rapid, standardized classification of cells based on pre-trained models [15]. The standard CellTypist workflow utilizes logistic regression classifiers optimized by stochastic gradient descent (SGD) to transfer cell type labels from reference models to query data [14] [4]. While the built-in models (e.g., Immune_All_Low.pkl and Immune_All_High.pkl) provide excellent starting points, particularly for immune cells, misclassification can occur when query cells are not adequately represented in the reference model's training data [14] [31]. This misclassification may stem from novel cell states not present in the reference, technical batch effects, or biological specificity (e.g., disease-specific cell states) that differ from the healthy references often used in pre-trained models [6] [31].
Addressing these misclassifications is not merely a technical exercise but fundamental to biological discovery. Custom models in CellTypist provide a powerful solution by enabling researchers to incorporate domain-specific knowledge, study-specific cell states, and disease-relevant populations into the classification framework [1]. This approach is particularly valuable for researchers studying specialized immune responses in disease contexts, developmental trajectories, or treatment effects, where standard immune cell classifications may lack the necessary resolution or relevance [31]. By building custom models, researchers can create tailored classification systems that reflect the biological complexity of their specific experimental systems while maintaining the advantages of automated, scalable annotation.
When to Consider Developing a Custom Model
Indicators of Needed Customization
Recognizing when to develop a custom model is crucial for accurate cell type annotation. The following indicators suggest that a custom model may be necessary:
Persistent novel cell states: When a significant population of cells (typically >5% of total cells) consistently receives low confidence scores (e.g., probability <0.5) or is classified as "unassigned" across multiple classification modes [6] [1]. This pattern suggests the presence of cell states not represented in existing reference models.
Biologically implausible annotations: The assignment of cell type labels that contradict established marker gene expression or expected biological context. For example, the classification of tissue-resident cells as circulating immune populations without supporting marker evidence indicates potential misclassification [31] [9].
Inconsistent subpopulation resolution: When the classification system lacks the resolution to distinguish functionally distinct subpopulations relevant to the research question. For instance, the inability to differentiate between activated and memory T cell subsets in an immunotherapy study would limit biological insights [14] [31].
Cross-tissue or cross-species applications: When working with tissues or species poorly represented in existing CellTypist models. While CellTypist focuses on human immune cells, applications in model organisms or non-immune tissues often require custom references [31].
Disease-specific cell states: When studying pathological conditions that generate unique cellular states not present in healthy reference atlases. This is particularly relevant in cancer, autoimmune, and infectious disease research [31].
Quantitative Assessment Framework
Systematically evaluate the need for a custom model using the following quantitative framework:
Table 1: Metrics for Assessing the Need for a Custom Model
| Assessment Metric | Calculation Method | Threshold for Custom Model | Interpretation |
|---|---|---|---|
| Unassigned Cell Rate | Percentage of cells with maximum probability <0.5 in 'prob match' mode [6] | >5% of total cells | Indicates substantial novel populations |
| Cross-Study Consistency | Concordance rate when applying multiple pre-trained models [31] | <70% agreement between models | Highlights reference-specific biases |
| Marker Gene Concordance | Percentage of cells where classifier label matches independent marker evidence [9] | <80% concordance | Suggests biological misclassification |
| Cluster Purity | Homogeneity of predicted labels within Leiden clusters [1] [9] | <75% purity in >10% of clusters | Reveals overlapping classifications |
Experimental Design for Custom Model Development
Reference Data Curation and Preprocessing
The foundation of an effective custom model is a comprehensively annotated reference dataset. The curation process should prioritize:
Data quality and annotation consistency: Select reference datasets with high-quality cell type annotations, preferably using standardized ontologies like the Cell Ontology [68]. Inconsistent labeling within the reference data will propagate to the custom model's performance.
Biological relevance: Ensure the reference encompasses the cell states relevant to your research question. For disease-focused studies, incorporate data from relevant pathological contexts rather than relying exclusively on healthy references [31].
Technical compatibility: When integrating multiple datasets to create a comprehensive reference, address batch effects using integration methods like Harmony, scVI, or Scanorama before model training [68]. The goal is biological diversity without technical artifacts.
The preprocessing of reference data must follow CellTypist's requirements for optimal performance:
Table 2: Reference Dataset Requirements for Custom Model Training
| Parameter | Requirement | Purpose | Quality Control |
|---|---|---|---|
| Cell Number | Minimum 1,000 cells per cell type [1] | Ensures robust parameter estimation | exclude cell types with <500 cells |
| Gene Coverage | Minimum 5,000 variable genes | Captures transcriptional diversity | Assess by genes/cell distribution |
| Annotation Granularity | Consistent hierarchy level | Prevents ambiguous classifications | Validate with marker genes |
| Technical Variance | Balanced protocol representation | Reduces platform-specific bias | Apply batch correction if needed |
Model Training Protocol
CellTypist provides a streamlined workflow for custom model training based on logistic regression with optional stochastic gradient descent (SGD) optimization for large datasets [14] [1]. The detailed training protocol consists of:
Step 1: Data Preparation
- Format the annotated reference data as an AnnData object with cell type labels in the
obsdataframe - Ensure the expression matrix is raw counts that will be normalized during preprocessing
- Verify that gene symbols are standardized and match the query dataset convention
Step 2: Feature Selection
- Enable feature selection during training to identify the most informative genes for classification [1]
- This step automatically identifies genes that contribute most to discriminating between cell types
- For specialized applications, pre-filter genes to focus on relevant gene sets (e.g., immune-specific genes)
Step 3: Model Training Configuration
Step 4: Model Validation
- Evaluate model performance through cross-validation within the training data
- Assess classification accuracy across cell types, identifying poorly performing classes
- Validate on held-out datasets or through independent marker expression analysis
The training process implements regularized logistic regression, with SGD optimization activating automatically for large datasets (>100,000 cells) using mini-batch training [14]. In this approach, cells are shuffled and divided into 1,000-cell batches, with training proceeding through 100 randomly sampled batches per epoch for 10-30 epochs [14] [15].
Model Evaluation and Iterative Refinement
Thorough evaluation is essential before deploying a custom model. The validation framework should include:
Cross-validation accuracy: Assess performance using k-fold cross-validation (typically k=5) to identify cell types with consistently poor classification [1]
Independent validation dataset: Test the model on completely held-out data not used during training, simulating real-world performance [31]
Marker gene concordance: Verify that predicted labels align with established marker gene expression in the validation data [9]
Comparison to existing models: Benchmark performance against relevant pre-trained CellTypist models to ensure meaningful improvement [31]
The iterative refinement process should focus on addressing identified weaknesses:
Implementation Workflow for Custom Models
Integrated Classification Strategy
An effective classification strategy combines custom and pre-trained models to leverage their respective strengths. The recommended workflow integrates both approaches:
Integrated Classification Strategy: This workflow outlines the decision process for implementing custom models, beginning with standard pre-trained models and proceeding to custom development only when necessary.
The integrated approach applies both pre-trained and custom models to query data, comparing results to identify consistent versus divergent classifications. Cells with concordant labels across approaches represent high-confidence annotations, while discordant classifications require additional investigation through marker gene expression or other orthogonal validation methods [31] [9].
Advanced Classification Modes
CellTypist offers multiple classification modes that can be strategically deployed based on the biological context:
Best match mode (default): Assigns each cell to the single most probable cell type [1]. This approach works well for homogeneous cell populations with clear distinctions between types.
Probability match mode: Allows multi-label classification where cells can be assigned to 0, 1, or multiple cell types based on a probability threshold (default: 0.5) [6]. This mode is particularly valuable for identifying:
- Intermediate or transitional cell states
- Cells with dual characteristics
- Novel cell types that don't reach threshold for any reference type
Majority Voting Refinement
The majority voting approach refines initial predictions by leveraging local neighborhood information [1] [8]. This process:
- Over-clusters the query data using Leiden clustering with a high resolution parameter
- Aggregates predictions within each subcluster
- Assigns consensus labels based on the most frequent prediction within each subcluster
This approach counters individual cell misclassifications by considering the transcriptional similarity of neighboring cells, often improving annotation consistency [8].
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Custom Model Development
| Reagent/Tool | Function | Specification Guidelines |
|---|---|---|
| Reference Datasets | Training data for custom models | Minimum 1,000 cells/cell type; Cell Ontology annotations preferred [68] |
| CellTypist Package | Core classification engine | Python 3.6+; requires scikit-learn, scanpy dependencies [4] |
| Scanpy | scRNA-seq data preprocessing | Required for normalization, clustering, and visualization [7] |
| Batch Correction Tools | Technical artifact mitigation | Harmony, scVI, or Scanorama for multi-dataset integration [68] |
| Marker Gene Databases | Validation resources | CellMarker, PanglaoDB, or literature-curated signatures [31] |
| Cell Ontology | Standardized annotation framework | Provides consistent cell type terminology across studies [68] |
Validation Framework and Quality Metrics
Orthogonal Validation Methods
Robust validation of custom models requires multiple orthogonal approaches:
Marker gene expression: Verify that predicted cell types express established marker genes not used in model training [9]. Create visualization overlays of key markers on UMAP projections of predicted labels.
Cross-dataset generalizability: Test the custom model on independent datasets from different studies or experimental conditions to assess robustness [31].
Cluster coherence analysis: Evaluate whether the custom model predictions produce coherent clusters in low-dimensional embeddings compared to pre-trained models [9].
Biological plausibility assessment: Ensure that the relative frequencies of predicted cell types align with biological expectations for the tissue or condition being studied.
Performance Benchmarking
Quantitatively benchmark custom model performance against relevant pre-trained models:
Table 4: Custom Model Performance Benchmarking Metrics
| Performance Metric | Calculation Method | Acceptance Threshold | Comparative Analysis |
|---|---|---|---|
| Cell Type F1-Score | Harmonic mean of precision/recall per cell type | >0.7 for all major types | Compare to pre-trained model baselines |
| Cross-Study Accuracy | Concordance when applied to independent data | >75% on relevant tissues | Assess generalizability beyond training data |
| Rare Cell Detection | Sensitivity for populations <5% abundance | >50% recall for biologically critical types | Evaluate clinical/research utility |
| Runtime Efficiency | Processing time per 10,000 cells | <5 minutes on standard workstation | Ensure practical applicability |
Systematic validation using this framework ensures that custom models provide genuine improvements over existing alternatives rather than simply overfitting to specific datasets or experimental conditions.
Custom CellTypist models represent a powerful approach for addressing the limitations of pre-trained classifiers in specialized research contexts. By following the structured framework presented here—from recognizing the need for custom models through systematic development and rigorous validation—researchers can create tailored classification systems that accurately capture the biological complexity of their specific experimental systems. The integrated strategy of combining custom and pre-trained models provides a robust foundation for cell type annotation that balances specificity with generalizability. As single-cell technologies continue to evolve and explore increasingly specialized biological questions, these custom approaches will become essential tools for extracting meaningful biological insights from complex scRNA-seq datasets.
Data normalization is a systematic process of structuring data to minimize redundancy, improve integrity, and ensure consistency. In the context of single-cell RNA sequencing (scRNA-seq) analysis with CellTypist, normalization ensures that input data is organized and reliable, forming a stable foundation for accurate automated cell type annotation [69] [70]. This process is crucial for researchers and drug development professionals who require trustworthy cellular composition data for downstream analysis and experimental validation.
For immune cell annotation research using CellTypist, proper data normalization directly impacts annotation reliability. Normalization eliminates inconsistencies that could propagate into broken data joins, misaligned reporting, and ultimately, erroneous biological interpretations [69]. By enforcing structural and semantic consistency, normalization ensures that CellTypist's logistic regression framework operates on stable, well-structured input data, leading to more accurate and reproducible cell type predictions [14].
Understanding Normalization Forms and Their Applications
Fundamental Normal Forms
Database normalization follows progressive guidelines called normal forms, which systematically reduce redundancy and prevent data anomalies [69] [71]. For scientific data management, including the organization of scRNA-seq metadata and experimental conditions, these forms provide a structured approach to data organization.
Table: Normal Forms and Their Applications in Research Data Management
| Normal Form | Core Requirement | Application in Research Data |
|---|---|---|
| First Normal Form (1NF) | Atomic values, no repeating groups | Ensuring single values per field in cell metadata tables |
| Second Normal Form (2NF) | No partial dependencies on composite primary keys | Structuring sample-condition relationships properly |
| Third Normal Form (3NF) | No transitive dependencies between non-key attributes | Separating experimental parameters from sample data |
First Normal Form (1NF)
1NF requires that each table column contains only atomic, indivisible values with no repeating groups [70] [71]. In scRNA-seq data management, this means instead of storing multiple marker genes in a single field as a comma-separated list, each gene should occupy a separate row or be stored in a dedicated table. This atomic structure ensures reliable querying and analysis [71].
Second Normal Form (2NF)
A table is in 2NF when it is already in 1NF and all non-key attributes are fully functionally dependent on the entire primary key [69] [70]. This is particularly relevant for tables with composite keys, such as those combining SampleID and CellID. In this case, attributes like donor information should depend only on SampleID, not the composite key, necessitating separation into related tables.
Third Normal Form (3NF)
3NF requires that a table is in 2NF and has no transitive dependencies, meaning non-key attributes must not depend on other non-key attributes [70] [71]. For example, if a cell metadata table contains both ZIP code and City fields, where City depends on ZIP code rather than directly on the primary key, this creates a transitive dependency that 3NF eliminates through table separation.
Normalization in Machine Learning Context
In machine learning, including CellTypist's framework, normalization refers to feature scaling rather than database structuring [69]. CellTypist employs logistic regression, potentially with SGD learning for large datasets, requiring properly scaled input features for optimal performance [14]. This transformation of gene expression values to a common scale ensures that highly expressed genes do not disproportionately influence the model's predictions.
Data Normalization Protocol for CellTypist Compatibility
Pre-normalization Data Assessment
Before normalization, comprehensively analyze your scRNA-seq data structure:
- Identify entities: Determine distinct objects in your data model (samples, cells, genes, experimental conditions) [70]
- Review data sources: Examine imported files for repeated fields, recurring patterns, and values that consistently appear together [70]
- Establish primary keys: Ensure each table has a unique identifier (SampleID, CellID) for maintaining record uniqueness and table relationships [70]
Step-by-Step Normalization Procedure
Table: Normalization Protocol for scRNA-seq Data
| Step | Procedure | Quality Control Check |
|---|---|---|
| 1. Apply 1NF | Ensure each field contains single values; create separate tables for multi-value attributes | Verify no comma-separated lists or arrays in fields |
| 2. Establish Relationships | Define logical connections between entities using foreign keys | Confirm valid references between tables; no orphaned records |
| 3. Implement 2NF | Remove partial dependencies by moving attributes dependent on part of composite keys | Check tables with composite keys for properly separated attributes |
| 4. Enforce 3NF | Eliminate transitive dependencies by creating separate tables for indirectly related attributes | Verify no non-key attributes depend on other non-key attributes |
| 5. Validate Structure | Test normalized schema with sample data; run common queries | Ensure structure supports all anticipated analytical queries |
Protocol Details
Step 1: Apply First Normal Form
- Convert all multi-value fields to single-value format
- Create junction tables for many-to-many relationships
- Ensure consistent data types across all records
Step 2: Establish Table Relationships
- Define one-to-many relationships between samples and cells
- Implement many-to-many relationships between cells and genes through junction tables
- Enforce referential integrity to maintain valid relationships
Step 3: Implement Second Normal Form
- Identify composite primary keys in analytical tables
- Remove attributes that depend on only part of composite keys
- Create separate tables for these attributes with proper relationships
Step 4: Enforce Third Normal Form
- Identify transitive dependencies where attributes depend on other non-key attributes
- Create lookup tables for commonly repeated values
- Establish foreign key relationships to maintain data integrity
Step 5: Validation and Testing
- Test with representative sample data
- Verify all required queries execute correctly
- Ensure data integrity during insertion, updates, and deletion
CellTypist-Specific Normalization Requirements
For optimal CellTypist performance, additional normalization steps are required:
- Expression value scaling: Apply appropriate feature scaling to gene expression data
- Metadata completeness: Ensure all required cellular metadata is present and properly structured
- Format compliance: Verify data format compatibility with CellTypist input requirements [14]
Experimental Protocols for Normalization Validation
Protocol 1: Data Integrity Verification
Purpose: To validate that normalization has properly maintained data integrity and relationships throughout the scRNA-seq dataset.
Materials:
- Normalized scRNA-seq dataset
- Computing environment with appropriate database management tools
- Data validation scripts
Procedure:
- Execute referential integrity tests to verify all foreign key relationships are valid
- Perform anomaly testing by simulating insertion, update, and deletion operations
- Validate that no redundant data exists across tables
- Verify that all business rules and constraints are properly enforced
- Document any integrity issues discovered and their resolutions
Quality Control: All integrity tests must pass before proceeding to CellTypist analysis. Any failures indicate required adjustments to the normalization structure.
Protocol 2: CellTypist Compatibility Testing
Purpose: To ensure normalized data is fully compatible with CellTypist's annotation framework and will produce reliable results.
Materials:
- Normalized scRNA-seq data
- CellTypist installation or access
- Benchmark dataset with known annotations (optional)
Procedure:
- Format normalized data according to CellTypist input specifications [14]
- Execute preliminary annotation run with subset of data
- Compare results with expected outcomes or benchmark data
- Verify all metadata is properly accessible during annotation process
- Validate that expression data is correctly scaled and normalized
Quality Control: CellTypist should execute without data format errors and produce biologically plausible annotations.
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for scRNA-seq Data Normalization and Analysis
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| CellTypist Platform | Automated cell type annotation using logistic regression models | Select appropriate immune cell models (e.g., ImmuneAllLow/High) based on research context [14] |
| Normalization Software | Implement database normalization and data structuring | Use Knack, Python Pandas, or R-based tools depending on technical requirements [69] [70] |
| Quality Control Tools | Validate data integrity and normalization effectiveness | Implement custom scripts or use data validation frameworks |
| Reference Datasets | Benchmark normalization and annotation performance | Use well-annotated public scRNA-seq datasets (e.g., PBMC data) [22] |
| LLM Integration Tools | Enhance annotation reliability through multi-model approaches | Implement LICT or similar frameworks for objective credibility evaluation [22] |
Advanced Normalization Strategies for Complex Research Data
Multi-Model Integration for Enhanced Reliability
For critical research applications, implement a multi-model integration strategy to improve annotation reliability. This approach leverages complementary strengths of multiple models to reduce uncertainty, particularly for low-heterogeneity cell populations that challenge individual models [22].
Procedure:
- Identify top-performing models for your specific research context
- Generate independent annotations using each model
- Integrate results using consensus approaches or credibility evaluation
- Resolve discrepancies through additional validation or expert consultation
Objective Credibility Evaluation Framework
Implement an objective framework to distinguish methodological discrepancies from intrinsic dataset limitations [22]:
- Marker gene retrieval: Query models for representative marker genes for each predicted cell type
- Expression pattern evaluation: Assess marker gene expression in corresponding cell clusters
- Credibility assessment: Classify annotations as reliable if >4 marker genes expressed in ≥80% of cluster cells
This framework provides greater confidence in identifying reliably annotated cell types for downstream analysis, overcoming limitations of relying solely on expert judgment.
Proper data normalization is fundamental for ensuring compatibility between scRNA-seq data and CellTypist's model expectations. By systematically applying normalization principles, researchers can significantly enhance the reliability of automated cell type annotations, particularly for immune cell research. The protocols and strategies outlined here provide a comprehensive framework for preparing data that meets CellTypist's requirements while maintaining the integrity and biological relevance of the underlying single-cell data. Implementation of these best practices will enable researchers and drug development professionals to generate more accurate, reproducible cellular annotations, thereby supporting robust downstream analysis and biological insights.
Troubleshooting installation and dependency conflicts
CellTypist is an automated cell type annotation tool for single-cell RNA-sequencing (scRNA-seq) datasets based on logistic regression classifiers optimised by the stochastic gradient descent algorithm [11]. It allows for cell prediction using either built-in models (with a current focus on immune sub-populations) or custom models, assisting researchers in accurately classifying different cell types and subtypes [11]. For immune cell annotation research, CellTypist provides specific models like "ImmuneAllLow" and "ImmuneAllHigh" which contain immune cell types collected from different tissues, with "Low" indicating low-hierarchy (high-resolution) cell types and subtypes and "High" indicating high-hierarchy (low-resolution) ones [14].
Despite its utility, researchers often encounter installation challenges and dependency conflicts when setting up CellTypist, which can hinder their ability to leverage this powerful annotation tool. This protocol provides comprehensive troubleshooting guidance to overcome these obstacles, ensuring researchers can successfully implement CellTypist in their immune cell annotation workflows.
Installation methods and quantitative comparison
Available installation options
CellTypist can be installed via two primary package managers, each with distinct characteristics and performance considerations:
pip Installation:
Conda Installation:
Comparative performance analysis
Table 1: Quantitative comparison of CellTypist installation methods
| Installation Method | Success Rate | Dependency Conflict Frequency | Recommended Python Version | Platform Support |
|---|---|---|---|---|
| pip | High | Low | 3.6+ | Linux, macOS, Windows |
| conda | Variable | High | 3.8, 3.9 | Linux, macOS |
Empirical evidence from multiple user reports indicates significant variability in installation success rates between these methods [72] [73]. Users have reported frequent dependency conflicts and environment solving failures with conda installations on both Ubuntu and macOS platforms, while pip installations generally proceed with fewer complications [72] [73]. One user specifically noted that after extensive troubleshooting with conda, including attempting downgraded versions of matplotlib, switching to pip installation resolved the issues immediately [72].
Common dependency conflicts and resolution protocols
Frequently problematic dependencies
Based on issue reports and community feedback, the following dependencies commonly cause conflicts during CellTypist installation:
- matplotlib: Version compatibility issues with other scientific Python packages
- scikit-learn: Particularly versions ≥1.3.0 requiring code adaptations [11]
- numpy: Matrix to array conversion requirements for prediction compatibility [11]
- pandas: Categorical output formatting issues in newer versions [11]
- leidenalg: Requirement for versions ≥0.9.0 [11]
Conflict resolution workflow
The following diagram illustrates the systematic troubleshooting approach for resolving CellTypist installation conflicts:
Step-by-step resolution protocol
Protocol 1: Dependency conflict resolution for CellTypist installation
Initial conda installation attempt
- Create a fresh conda environment:
conda create -n celltypist python=3.9 - Activate the environment:
conda activate celltypist - Attempt conda installation:
conda install -c bioconda -c conda-forge celltypist
- Create a fresh conda environment:
Conflict identification and resolution
Alternative pip installation method
- Within the same conda environment, switch to pip installation:
pip install celltypist - Alternatively, create a fresh environment and use pip exclusively without conda
- Within the same conda environment, switch to pip installation:
Installation verification
- Launch Python:
python - Import CellTypist:
import celltypist - If import succeeds without errors, installation is successful [72]
- Launch Python:
Advanced troubleshooting (if required)
Research reagent solutions for CellTypist implementation
Table 2: Key research reagent solutions for CellTypist workflows
| Resource Type | Specific Solution | Function/Purpose | Implementation in Immune Research |
|---|---|---|---|
| Computational Environment | Python 3.6+ | Base programming language for CellTypist | Required for all immune cell annotation tasks |
| Package Manager | pip | Dependency management and installation | Preferred method based on success rates [72] |
| Container Solution | Docker/Singularity | Environment consistency across systems | Alternative when dependency issues persist [11] |
| GPU Acceleration | NVIDIA GPU with CUDA | Speed up training and over-clustering | Optional for large immune datasets (≥1.7.0) [11] |
| Reference Models | ImmuneAllLow / ImmuneAllHigh | Pre-trained classifiers for immune cells | Foundation for automated immune cell annotation [14] |
Experimental annotation workflow for immune cells
Complete immune cell annotation protocol
Protocol 2: Comprehensive immune cell annotation using CellTypist
Data preparation and environment setup
- Format scRNA-seq data as CSV or H5AD files [4]
- Ensure proper gene expression matrix formatting (cells × genes)
- Verify CellTypist installation following Protocol 1
Model selection for immune cells
- Select appropriate immune reference model based on resolution needs:
Immune_All_Low: For high-resolution immune subpopulation discriminationImmune_All_High: For broad immune cell categorization [14]
- Consider custom model training for specialized immune applications
- Select appropriate immune reference model based on resolution needs:
Cell type prediction execution
Result interpretation and validation
- Compare immune subset proportions across experimental conditions
- Validate annotations using known immune marker genes
- Employ majority voting to refine immune subtype calls [4]
Advanced GPU acceleration protocol
Protocol 3: GPU-accelerated annotation for large immune datasets
GPU environment configuration
- Install CellTypist version ≥1.7.0 for GPU support [11]
- Ensure CUDA-compatible NVIDIA GPU with appropriate drivers
- Install GPU dependencies via conda or pip
GPU-enabled execution
- Use
--gpuflag in command line interface [11] - For Python API, enable GPU acceleration during model training and prediction
- Monitor GPU memory usage for large immune datasets (>100,000 cells)
- Use
Performance optimization
- Batch process extremely large datasets using chunking
- Utilize over-clustering with GPU acceleration [11]
- Implement appropriate data preprocessing to maximize GPU efficiency
Validation and quality control measures
Annotation reliability assessment
To ensure robust immune cell annotation results, implement the following quality control measures:
- Cross-validation with manual annotation: Compare CellTypist results with expert immune cell identification using canonical markers [31]
- Marker gene expression verification: Validate predicted immune cell types by checking expression of characteristic markers (e.g., CD3E for T cells, CD19 for B cells)
- Majority voting implementation: Use CellTypist's majority voting feature to improve annotation accuracy for immune subtypes [4]
- Independent dataset validation: Verify annotation consistency using technical or biological replicates
Performance benchmarking
Studies evaluating automated annotation tools report that CellTypist predictions can achieve approximately 65.4% agreement with manual annotations in complex immune datasets [31]. However, performance varies based on dataset quality, reference model appropriateness, and immune cell type rarity. For immune cell annotation, the tool particularly excels in identifying common lymphoid and myeloid lineages while potentially facing challenges with rare immune subsets or activated states requiring specialized reference models.
Performance Optimization for Cross-Dataset Label Transfer
CellTypist is an automated cell type annotation tool for single-cell RNA sequencing (scRNA-seq) data, leveraging logistic regression classifiers optimized by stochastic gradient descent (SGD) [1] [2]. Its framework is particularly valuable for immune cell annotation research, enabling accurate classification of immune cell types and subtypes across tissues and datasets. The tool allows researchers to perform predictions using either built-in models (with a focus on immune sub-populations) or custom-trained models, facilitating cross-dataset label transfer in diverse research contexts [1] [2].
A key strength of CellTypist is its training on comprehensive immune cell references encompassing multiple tissues, creating an organ-agnostic classification system ideal for immune compartments shared across different tissues [2]. Performance evaluations demonstrate that CellTypist achieves precision, recall, and F1-scores of approximately 0.9 for immune cell classification at both high- and low-hierarchy levels, outperforming or matching other label-transfer methods with minimal computational cost [2].
Performance Optimization Strategies
Model Selection and Configuration
Optimal model selection is fundamental for performance in cross-dataset label transfer. CellTypist provides multiple built-in models specifically designed for immune cell annotation, with selection guidance based on the research question and desired resolution [14].
Table 1: CellTypist Model Selection for Immune Cell Annotation
| Model Name | Resolution | Cell Types | Use Case | Performance Considerations |
|---|---|---|---|---|
| ImmuneAllLow | Low-hierarchy (High-resolution) | 91 cell types and subtypes | Fine-grained immune sub-population discrimination | Distinguishes CD4+ T cell subsets (helper, regulatory, cytotoxic) and CD8+ T cell subpopulations including MAIT cells [2] |
| ImmuneAllHigh | High-hierarchy (Low-resolution) | 32 cell types | Major immune population identification | Clearly resolves monocytes from macrophages and identifies progenitor populations [2] |
| Custom-trained models | Variable | User-defined | Dataset-specific requirements | Can be optimized for specific tissue contexts or rare cell populations |
For optimal performance, the Immune_All_Low model is recommended when seeking high-resolution immune cell annotation, as it can distinguish 43 specific immune subtypes including T cell subsets, B cell states, ILCs, and dendritic cell populations (DC1, DC2, and migDCs) [2]. The model choice should align with the annotation granularity required for the specific research question.
Data Preprocessing Optimization
Proper data preprocessing significantly impacts CellTypist performance. The input data should be provided as a raw count matrix (reads or UMIs) in a cell-by-gene format [1]. For gene-by-cell formatted data, researchers must set transpose_input = True during annotation [1].
Critical preprocessing considerations include:
- Inclusion of non-expressed genes: These provide negative transcriptomic signatures important for model comparison [1]
- Data normalization: When using the
to_adatafunction, expression matrices are automatically log1p normalized to 10,000 counts per cell [1] - Quality control: Implementation of stringent quality control metrics as demonstrated in large-scale cross-tissue studies, which eliminated approximately 8% of cells in a 357,211-cell dataset [2]
- Memory optimization: Setting
with_mean = Falseduring model training reduces memory usage when working with sparse matrices, with a potential slight decrease in model performance [13]
For large-scale analyses exceeding 100,000 cells, the mini_batch = True parameter activates mini-batch training, improving training efficiency without significant performance loss [13].
Computational Efficiency Enhancements
Computational performance optimization is crucial when working with large-scale datasets common in immune cell atlas projects. CellTypist offers several parameters to enhance processing efficiency:
Table 2: Computational Performance Parameters for Large Datasets
| Parameter | Setting for Large Datasets | Impact on Performance | Use Case |
|---|---|---|---|
use_SGD |
True |
Enables stochastic gradient descent learning | Datasets >50,000 cells [13] |
mini_batch |
True |
Implements mini-batch training | Datasets >100,000 cells [13] |
batch_size |
1000 (default) | Cells per batch in mini-batch training | Balances memory usage and performance |
batch_number |
100 (default) | Number of batches per epoch | Large-scale cross-dataset training |
epochs |
10 (default) | Training iterations | ~1,000,000 training cells observed with default settings [13] |
n_jobs |
-1 (all CPUs) | Parallel processing | Maximizes computational efficiency on multi-core systems [13] |
The SGD optimizer with mini-batch training is particularly effective for large datasets, as it processes data in sequential batches (1,000 cells per batch) across multiple epochs, significantly reducing memory requirements while maintaining model accuracy [2] [13].
Experimental Protocols for Cross-Dataset Label Transfer
Basic Label Transfer Workflow
The foundational protocol for cross-dataset label transfer using CellTypist involves these critical steps:
Step 1: Installation and Setup
Step 2: Model Selection and Download
Step 3: Data Loading and Preparation
Step 4: Prediction Execution
Step 5: Result Extraction and Export
Advanced Optimization Protocol for Large Datasets
For datasets exceeding 50,000 cells or multi-dataset integration projects, advanced optimization protocols are recommended:
Protocol for Large-scale Immune Cell Annotation:
Custom Model Training for Enhanced Performance:
Validation and Quality Assessment:
Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for CellTypist Workflows
| Resource Category | Specific Tool/Resource | Function in Workflow | Performance Impact |
|---|---|---|---|
| Pre-trained Models | ImmuneAllLow.pkl | High-resolution immune cell annotation | Provides ~0.9 F1-score for immune cell classification [2] |
| Pre-trained Models | ImmuneAllHigh.pkl | Major immune population annotation | Resolves monocytes/macrophages and progenitor populations [2] |
| Data Formats | .h5ad (AnnData) | Efficient storage of large single-cell datasets | Enables faster I/O operations for large datasets |
| Data Formats | .mtx with gene/cell files | Memory-efficient matrix storage | Suitable for extremely large datasets (>1M cells) |
| Computational Resources | Multiple CPU cores (n_jobs = -1) | Parallel processing during training | Reduces computation time for large-scale analysis [13] |
| Validation Tools | Marker gene expression plots | Quality assessment of predictions | Verifies biological consistency of annotations |
| Validation Tools | Probability matrices | Confidence assessment of cell type calls | Identifies low-confidence assignments requiring manual review |
Troubleshooting and Quality Assessment
Addressing Common Performance Issues
Low Annotation Confidence:
- Symptoms: Low probability scores across cell populations
- Solutions:
- Switch to
mode = 'prob match'with adjustedp_thres(0.3-0.5) - Verify reference model compatibility with query data
- Check data normalization (should be log1p normalized to 10,000 counts/cell)
- Switch to
Computational Resource Limitations:
- Symptoms: Memory errors or excessive processing time
- Solutions:
- Activate
use_SGD = Trueandmini_batch = Truefor datasets >50,000 cells - Set
with_mean = Falseto reduce memory footprint - Utilize
n_jobs = -1for parallel processing across all available CPUs
- Activate
Poor Cross-Dataset Generalization:
- Symptoms: Inconsistent annotations across similar cell types
- Solutions:
- Train custom model with
balance_cell_type = Trueto address rare populations - Enable
feature_selection = Trueto focus on informative genes - Verify batch effects are not dominating biological signals
- Train custom model with
Quality Metrics and Validation
Performance optimization should be validated using both computational and biological metrics:
Computational Performance Metrics:
- Processing time per 10,000 cells
- Memory usage peaks during annotation
- CPU utilization efficiency
Biological Validation Metrics:
- Expression of canonical immune marker genes in annotated populations
- Consistency with manual annotations (when available)
- Cross-validation with independent classification methods
- Confidence scores distribution across cell populations
The optimization approaches outlined here enable researchers to efficiently perform cross-dataset label transfer for immune cell annotation while maintaining biological accuracy and computational efficiency, supporting robust and reproducible single-cell research in immunology and drug development.
Common Pitfalls in Data Formatting and Transpose Errors
CellTypist requires specific data formats to function optimally for immune cell annotation. The tool expects a raw count matrix as input, which can be provided in either cell-by-gene or gene-by-cell orientation [1]. For proper classification, this matrix should include both expressed and non-expressed genes, as the negative transcriptomic signatures provide important contrasting information against the model's reference patterns [1].
A critical preprocessing step involves normalization and transformation where the raw counts are normalized to 10,000 counts per cell followed by log1p transformation [8]. This standardized processing ensures compatibility with the CellTypist models, which were trained on similarly processed data. When these formatting requirements are not meticulously followed, researchers encounter predictable but avoidable errors that compromise annotation accuracy.
Table 1: Essential Input Data Specifications for CellTypist
| Parameter | Requirement | Consequence of Deviation |
|---|---|---|
| Matrix type | Raw counts (reads or UMIs) | Incorrect normalization leads to poor model performance |
| Normalization | Total count normalization to 10,000 per cell | Scaling artifacts and inaccurate cell type probabilities |
| Transformation | log1p applied after normalization | Non-linear distortion of gene expression relationships |
| Gene coverage | All genes (including zeros) | Loss of negative signature information important for classification |
| Data orientation | Configurable (cell-by-gene or gene-by-cell) | Transpose errors causing complete misclassification |
Data Formatting Pitfalls and Solutions
Matrix Orientation and Transpose Errors
The most common formatting error in CellTypist implementation involves incorrect matrix orientation. By default, CellTypist expects a cell-by-gene matrix with cells as rows and gene symbols as columns [1]. When data is provided in gene-by-cell format without proper parameter adjustment, the tool attempts to classify genes as cells, yielding nonsensical results.
To prevent transpose errors, explicitly specify the orientation using the transpose_input parameter:
For MTX format files, which are commonly output by droplet-based sequencing technologies, additional file parameters must be specified to correctly annotate rows and columns:
Normalization and Transformation Issues
Improper normalization represents another frequent pitfall. CellTypist models are trained on data normalized to 10,000 counts per cell with log1p transformation [8]. When researchers input raw counts without normalization or use alternative normalization schemes, the gene expression values fall outside the expected range of the trained models, reducing prediction accuracy.
The correct preprocessing workflow should follow this sequence:
- Begin with raw UMI counts or read counts
- Normalize total counts per cell to 10,000 using
scanpy.pp.normalize_total(target_sum=1e4) - Apply log1p transformation using
scanpy.pp.log1p() - Use this normalized data for CellTypist prediction
Table 2: Troubleshooting Common Data Formatting Errors
| Error Symptom | Likely Cause | Solution |
|---|---|---|
| Dimension mismatch errors | Matrix orientation incorrect | Use transpose_input=True for gene-by-cell data |
| All cells classified as "Unassigned" | Improper normalization | Verify normalization to 10,000 counts/cell + log1p |
| Low confidence scores across all predictions | Incorrect count matrix type | Use raw counts (not already normalized data) |
| Model features not found in data | Gene symbol mismatch | Standardize gene symbols to match model requirements |
| Inconsistent results between runs | Non-expressed genes filtered | Retain zeros in input matrix for negative signatures |
Experimental Protocol for Data Preparation
Standardized Workflow for Immune Cell Annotation
The following protocol ensures properly formatted input for CellTypist, minimizing formatting and transpose errors in immune cell annotation research:
Step 1: Data Export and Formatting
- Export count data from your single-cell analysis tool (Seurat, Scanpy, etc.) as a raw count matrix
- Ensure gene symbols are in the same format as the CellTypist model (usually HGNC symbols)
- Preserve all genes in the matrix, including those with zero counts in some cells
- Save the matrix in a compatible format (.csv, .tsv, .txt, .mtx, or .h5ad)
Step 2: Matrix Orientation Verification
- Visually inspect the matrix dimensions: cells should be rows, genes should be columns for default orientation
- For .csv files, the first column should contain cell barcodes and first row should contain gene symbols
- Confirm matrix orientation matches your
transpose_inputparameter setting
Step 3: Normalization and Transformation
- Implement total count normalization to 10,000 counts per cell
- Apply log1p transformation to the normalized data
- For .h5ad files, store the normalized data in the appropriate layer
- For online CellTypist use, upload the normalized and transformed data
Step 4: Model Compatibility Check
- Compare the genes in your dataset with the features in your selected CellTypist model
- Verify that a sufficient percentage of model features are present in your data
- For immune cell annotation, select the appropriate model (e.g.,
Immune_All_Low.pklfor broad immune populations)
Step 5: CellTypist Execution
- Run CellTypist with the appropriate
transpose_inputparameter - For initial exploration, use the
mode = 'best match'parameter - For difficult classifications, consider
mode = 'prob match'with a custom probability threshold
Validation and Quality Control
After annotation, implement these quality control measures:
- Check the distribution of confidence scores across cell predictions
- Verify that known immune cell marker genes align with predicted cell types
- Compare automated annotations with manual cluster-based annotation when possible
- Use UMAP visualization to assess the coherence of predicted cell type groupings
Figure 1: Data formatting workflow for CellTypist - This diagram illustrates the critical decision points for proper data preparation, highlighting how to handle different matrix orientations to avoid transpose errors.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Resource | Type | Function in Cell Typing |
|---|---|---|
| CellTypist Python package | Software tool | Automated cell type annotation using logistic regression classifiers [11] |
| ImmuneAllLow.pkl model | Pre-trained model | Broad immune cell classification with 1,000 feature genes [1] |
| ImmuneAllHigh.pkl model | Pre-trained model | Detailed immune cell annotation with 2,000 feature genes [1] |
| Scanpy | Python package | Single-cell analysis including normalization and preprocessing [54] |
| 10x Genomics Cell Ranger | Analysis pipeline | Raw data processing from sequencing outputs to count matrices |
| PBMC reference datasets | Reference data | Benchmarking and validation of immune cell typing performance [9] |
| Custom model training function | Software feature | Creating dataset-specific classifiers using celltypist.train() [1] |
Ensuring Accuracy: Validation Strategies and Comparative Analysis
Within immune cell annotation research, ensuring consistency and biological accuracy is paramount. The Cell Ontology (CL) serves as a critical gold standard, providing a controlled, structured vocabulary of canonical cell types with defined hierarchical relationships [74] [75]. For researchers using CellTypist, a automated cell type annotation tool for scRNA-seq data [4], validation against the CL is not merely a final check but a fundamental step to verify that computationally derived labels correspond to biologically meaningful entities. This protocol details how to perform this alignment and verification, thereby strengthening the credibility of CellTypist's annotations and facilitating integration with other biologically grounded resources. Adherence to this framework is essential for robust, reproducible, and interpretable results in immunology and drug development.
Background and Significance
The Cell Ontology is a candidate ontology within the OBO Foundry, specifically designed to represent in vivo cell types across vertebrates [75]. Its value lies in a computable structure that defines cell types not only by specific marker genes but also by their anatomical location and developmental lineage. This allows for reasoning about relationships between cell types; for instance, a CD8-positive, alpha-beta T cell is a subclass of T cell [75]. Relying solely on gene expression patterns for annotation can lead to labels that are dataset-specific or lack biological context. Aligning CellTypist's output with the CL grounds predictions in established community knowledge, helps identify potentially novel cell types not yet in the ontology, and ensures that annotations are consistent with those used by major functional genomics consortia like FANTOM5 and ENCODE [75]. Furthermore, the CL's hierarchy allows for the evaluation of annotation errors by measuring the ontological distance between misclassified cell types, a more biologically informed metric than simple accuracy [76].
Workflow for Cell Ontology Alignment and Verification
The following diagram illustrates the end-to-end process for validating CellTypist annotations against the Cell Ontology.
Essential Toolkit for Cell Ontology Validation
The table below summarizes the key resources required to execute the validation protocol.
Table 1: Research Reagent Solutions for CL Validation
| Item Name | Function / Description | Source / Availability |
|---|---|---|
| CellTypist | Python-based tool for automated cell type annotation using logistic regression models. Provides pre-trained models and allows for custom training [4]. | celltypist.org; Available via PyPI (pip install celltypist) or Bioconda [4]. |
| Cell Ontology (CL) | The authoritative, structured vocabulary of cell types against which annotations are validated. Provides the hierarchical relationships and definitions [74] [75]. | OBO Foundry; Available in OWL format from ontobee.org or the CL GitHub repository. |
| OnClass Algorithm | A reference algorithm demonstrating the use of the CL graph and text descriptions to classify cells, including to unseen cell types. Serves as a conceptual benchmark [74]. | Python package; Methodology described in Nature Communications [74]. |
| CytoPheno Tool | An algorithm and tool that automates the mapping of marker gene profiles to standardized Cell Ontology terms, providing a practical validation pipeline [77]. | R Shiny application; Publicly available on GitHub. |
| scGraph-OntoRWR Metric | A novel evaluation metric that measures the consistency between cell-type relationships learned by a model and the known relationships in the Cell Ontology [76]. | Implementation referenced in benchmarking studies of single-cell foundation models [76]. |
Core Alignment and Verification Protocols
Protocol 1: Automated Lexical Mapping of Annotations
This protocol focuses on the initial alignment of CellTypist-generated labels with the correct terms in the Cell Ontology.
- Input Preparation: Execute CellTypist on your query scRNA-seq dataset. The output (
predictions.predicted_labelsfrom the CellTypist Python API) is a list of free-text cell type labels [4]. - Term Extraction: Parse the CellTypist output to create a unique list of predicted cell type strings.
- Lexical Matching: For each unique label, perform string matching against the official labels and synonyms in the current Cell Ontology (e.g.,
CD4-positive, alpha-beta T cell). Tools like CytoPheno implement this step, standardizing input names before matching [77]. - Similarity Scoring: For labels without an exact match, use natural language processing (NLP) to compute text-embedding similarities (e.g., cosine similarity) between the predicted label and all CL term descriptions. OnClass uses this approach to map free-text annotations to the CL [74].
- Output: Generate a mapping file linking each original CellTypist label to its best-matched CL term ID (e.g.,
CL:0000798forgamma-delta T cell) and the confidence score of the match.
Protocol 2: Hierarchical Consistency and Error Validation
This protocol uses the hierarchical structure of the CL to biologically validate the coherence of CellTypist's predictions.
- Hierarchy Import: Load the Cell Ontology, representing its "is_a" relationships as a directed acyclic graph.
- Annotation Projection: Project the verified annotations from Protocol 1 onto the CL graph. Each cell is associated with a specific node (term) in the graph.
- Similarity Calculation: For a given analysis (e.g., cross-dataset validation, benchmarking), calculate two similarity measures for cell types:
- Gene Expression Similarity: The correlation of average gene expression profiles.
- CL Graph-based Similarity: The cosine similarity between random walk with restart distributions on the CL graph, which encodes ontological proximity [74].
- Consistency Check: Assess the correlation between gene expression similarity and CL graph-based similarity. A strong positive correlation (e.g., ~0.65-0.93 as reported in foundational studies) indicates that CellTypist's predictions respect the biological relationships encoded in the CL [74].
- Error Severity Assessment: When misclassifications occur, use the Lowest Common Ancestor Distance (LCAD) metric. Instead of treating all errors equally, LCAD quantifies the ontological proximity between the predicted and true cell type. A small LCAD indicates a less severe error (e.g., confusing two T cell subtypes) [76].
Quantitative Validation and Benchmarking
When benchmarking CellTypist against other methods or a ground truth, incorporate CL-derived metrics for a biologically nuanced assessment.
Table 2: Key Metrics for Quantitative Validation Against Cell Ontology
| Metric Name | Description | Interpretation | Reported Performance |
|---|---|---|---|
| CL Alignment Score | Correlation between gene expression-based cell type similarity and Cell Ontology graph-based similarity. | Higher scores (closer to 1.0) indicate predictions are more consistent with known biology. | Foundational studies report correlations of 0.65 (lung) and 0.93 (pancreas) [74]. |
| Lowest Common Ancestor Distance (LCAD) | Measures the ontological distance between misclassified cell types and their true label in the CL hierarchy. | Lower LCAD values indicate less severe, more biologically plausible errors. | Used in benchmarking single-cell foundation models as a knowledge-based metric [76]. |
| Accuracy on Unseen Cell Types | The model's performance (e.g., AUROC) on cell types not present in the training data, enabled by the CL graph. | Demonstrates the power of using the CL to generalize beyond the training set. | OnClass achieved an AUROC of 0.87 vs. <0.67 for other methods when 70% of types were unseen [74]. |
| scGraph-OntoRWR | A metric that uses random walk with restart on the CL graph to evaluate the intrinsic biological knowledge in a model's latent space. | Higher scores indicate the model's internal representations better reflect ontological relationships. | A novel metric applied in the evaluation of single-cell foundation models [76]. |
Advanced Integration and Application
For complex immunology studies, particularly those involving T cell subtyping, a more advanced integrated workflow is recommended. The following diagram details this process, which combines automated annotation with hierarchical validation.
Workflow Description:
- Initial Annotation: Process T cell data with a high-resolution CellTypist model (e.g.,
Immune_All_Low) [14]. - CL Validation: Execute Protocols 1 and 2 to map and validate the initial annotations.
- Hierarchical Refinement: Employ a tool like STCAT, which uses a hierarchical model to first classify major lineages (e.g., CD4+ vs. CD8+) before fine-grained subtypes, improving accuracy by up to 28% compared to flat classification [25].
- Expert-Guided Curation: Use the CL's relationships to guide manual inspection. For example, if a cluster is annotated as
T helper 17 cell, an expert can verify the expected presence of marker genes like RORC and absence of markers for closely related subtypes likeT follicular helper cell, which are siblings in the CL hierarchy. - Atlas Generation: The final output is a verified cell atlas where annotations are consistent with the CL, enabling reliable cross-dataset comparison and biological insight discovery, such as identifying the enrichment of Th17 cells in late-stage lung cancer [25].
Comparing CellTypist Performance with Manual Annotation Approaches
Cell type annotation represents a critical step in single-cell RNA sequencing (scRNA-seq) analysis, with traditional manual approaches and emerging automated methods each presenting distinct advantages and limitations. This application note systematically evaluates CellTypist, an automated cell type annotation tool, against conventional manual annotation methodologies. We provide quantitative performance comparisons across diverse biological contexts, detailed experimental protocols for implementation, and practical guidance for researchers conducting immune cell annotation studies. Our analysis demonstrates that CellTypist achieves annotation accuracy comparable to manual expert annotation while significantly reducing processing time and subjective bias, particularly for well-characterized immune cell populations.
Manual Annotation
Traditional manual annotation relies on expert knowledge to interpret cluster-specific gene expression patterns against established marker genes from literature or databases [31]. This approach involves clustering cells into groups with similar expression profiles, identifying upregulated genes in each cluster through differential expression analysis, and manually comparing these genes to known canonical markers [31]. While offering complete researcher control and potentially high reliability when meticulously performed, manual annotation suffers from several limitations: substantial time investment, dependency on accurate clustering, inter-annotator variability, and inconsistent use of standardized ontologies [31].
Automated Annotation with CellTypist
CellTypist represents a reference-based automated annotation approach that employs logistic regression classifiers optimized by stochastic gradient descent algorithm [2] [11] [1]. This machine learning framework enables rapid cell type prediction without requiring prior clustering, leveraging pre-trained models built from extensively curated multi-tissue reference datasets [2]. CellTypist's current models focus particularly on immune cell sub-populations, providing both high-hierarchy (major cell types) and low-hierarchy (fine-grained subtypes) classification capabilities [2] [17].
Performance Comparison
Quantitative Performance Metrics
Table 1: Performance Comparison Across Annotation Methods
| Metric | Manual Annotation | CellTypist | LLM-Based Methods (LICT) |
|---|---|---|---|
| Accuracy with PBMCs | Gold standard | 65.4% match to manual annotations [31] | 90.3% match (9.7% mismatch) [22] |
| Processing Time | Hours to days | Minutes to hours [31] | Variable (model-dependent) |
| Inter-annotator Consistency | Variable/subjective | High/objective [2] | Objective framework [22] |
| Dependence on Reference | Literature markers | Pre-trained models [1] | No reference required [22] |
| Handling of Novel Cell Types | Flexible but laborious | Limited to model vocabulary | Potential for identification [22] |
| Low-heterogeneity Datasets | Expert interpretation | Challenges with fine distinctions | 43.8-48.5% match rates [22] |
Independent benchmarking using the Asian Immune Diversity Atlas (AIDA) v2 dataset demonstrated that CellTypist predictions matched author-provided manual annotations for 65.4% of cells (82,802 of 126,546 cells) [31]. Performance varies significantly based on the similarity between query data and the reference model's training data.
Performance Across Biological Contexts
Table 2: Performance Across Dataset Types
| Dataset Type | Manual Annotation Reliability | CellTypist Performance | Context Notes |
|---|---|---|---|
| High-heterogeneity (PBMCs) | Established | High accuracy for major populations [31] | Well-characterized immune cells |
| High-heterogeneity (Gastric Cancer) | Established | 69.4% full match rate [22] | Diverse tumor microenvironment |
| Low-heterogeneity (Embryonic Cells) | Expert-dependent | 48.5% full match rate [22] | Developing systems with subtle distinctions |
| Low-heterogeneity (Stromal Cells) | Challenging | 43.8% full match rate [22] | Finely resolved subtypes problematic |
| Cross-tissue Immune Cells | Consistent curation needed | Precision ~0.9 for classification [2] | Multi-tissue integration strength |
Recent advancements in large language model (LLM)-based approaches such as LICT (Large Language Model-based Identifier for Cell Types) demonstrate the evolving landscape of automated annotation, achieving mismatch rates as low as 9.7% for PBMC datasets through multi-model integration strategies [22]. However, CellTypist remains particularly valuable for immune cell annotation due to its specialized training on extensively curated immune cell references [2].
Experimental Protocols
Manual Annotation Protocol
Step-by-Step Procedure:
Data Preprocessing: Perform quality control to remove low-quality cells and genes, followed by normalization and scaling of the expression matrix.
Clustering: Reduce dimensionality using principal component analysis (PCA) and project cells into low-dimensional space using UMAP or t-SNE. Apply graph-based clustering algorithms (e.g., Leiden, Louvain) to identify cell communities.
Differential Expression Analysis: For each cluster, identify significantly upregulated genes compared to all other cells using appropriate statistical tests (e.g., Wilcoxon rank-sum test, MAST).
Marker Gene Consultation: Compare upregulated genes against established marker databases:
- CellMarker: Curated resource of cell markers across multiple tissues and species
- PanglaoDB: Specialized database focusing on scRNA-seq markers
- Literature: Primary research publications for cell-type-specific signatures
Annotation Assignment: Assign cell type identities to each cluster based on the overlap between cluster-specific markers and known cell type signatures.
Validation: Visually confirm annotation accuracy by examining expression patterns of canonical markers across clusters using violin plots, feature plots, or dot plots.
CellTypist Annotation Protocol
Step-by-Step Procedure:
Installation and Setup:
Data Preparation:
- Format input data as a raw count matrix with cells as rows and genes as columns
- Supported formats:
.txt,.csv,.tsv,.tab,.mtx, or.mtx.gz - Ensure gene symbols are provided as features
Model Selection:
Cell Type Prediction:
Result Refinement with Majority Voting:
Result Interpretation and Export:
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool/Resource | Type | Function | Application Context |
|---|---|---|---|
| CellTypist Python Package | Software | Automated cell type annotation | Primary classification tool [11] |
| Pre-trained Models | Reference Data | Immune cell classification basis | Model selection based on tissue type [1] |
| Scanpy | Software | scRNA-seq preprocessing and clustering | Essential for manual annotation workflow [8] |
| Seurat | Software | scRNA-seq analysis suite | Alternative to Scanpy ecosystem [34] |
| CellMarker Database | Reference | Curated cell marker repository | Manual annotation validation [31] |
| PanglaoDB | Reference | scRNA-seq marker database | Manual annotation support [31] |
| Cell Ontology | Standard | Structured cell type ontology | Standardized terminology [17] |
| AIDA Dataset | Benchmark Data | Asian Immune Diversity Atlas | Performance validation [31] |
Application Notes for Immune Cell Research
Strategic Implementation
For immune cell annotation research, we recommend a hybrid approach that leverages the respective strengths of both methods:
Primary Annotation with CellTypist: Utilize CellTypist for initial, rapid annotation of well-established immune cell populations (T cells, B cells, monocytes, etc.).
Expert Validation and Refinement: Employ manual annotation to validate predictions, resolve discrepancies, and identify novel or ambiguous populations that may require additional investigation.
Quality Assessment: Implement CellTypist's built-in confidence scores and probability matrices to identify low-confidence predictions that warrant manual review.
Optimization Guidelines
Model Selection: Choose models based on biological context; ImmuneAllLow.pkl serves as a robust starting point for general immune cell annotation [8] [1].
Data Compatibility: Ensure query data processing (normalization, gene naming) aligns with model training specifications to maximize performance.
Majority Voting: Consistently enable majority voting to improve annotation consistency, particularly for heterogeneous cell populations [8].
Cross-Validation: For critical applications, validate CellTypist predictions against a subset of manually annotated cells to establish method reliability for specific experimental contexts.
CellTypist provides a standardized, efficient, and scalable solution for immune cell annotation that demonstrates performance comparable to manual approaches for well-characterized cell types. While manual annotation remains essential for novel cell type discovery and complex biological contexts, CellTypist significantly accelerates analysis throughput and reduces subjective bias. The integration of both approaches through a hybrid framework offers the most robust strategy for comprehensive immune cell characterization in scRNA-seq studies, particularly valuable for drug development professionals requiring reproducible, standardized cell type annotation across multiple experiments and conditions.
Benchmarking against other automated annotation tools
Automated cell type annotation has become an essential step in single-cell RNA sequencing (scRNA-seq) analysis, addressing critical challenges in reproducibility, scalability, and accuracy faced by manual annotation approaches. CellTypist represents a machine learning-based solution specifically designed for precise immune cell classification across diverse tissue contexts. This tool employs logistic regression classifiers optimized by stochastic gradient descent learning to enable rapid and accurate cell type prediction [2] [1]. As single-cell technologies evolve and datasets expand exponentially, robust benchmarking of automated annotation tools becomes increasingly crucial for ensuring biological validity in downstream analyses.
The development of CellTypist was motivated by the limitations of existing annotation methods when applied to complex multi-tissue immune cell datasets. Traditional approaches often rely on manual cluster annotation using established marker genes, a process that introduces subjectivity and suffers from limited reproducibility across research groups [78]. CellTypist addresses these challenges through a pan-tissue immune database and automated classification pipeline that systematically resolves immune cell heterogeneity across tissues [2]. This protocol details comprehensive benchmarking methodologies to objectively evaluate CellTypist against alternative approaches, providing researchers with practical guidance for implementation within immune cell annotation workflows.
Performance comparison of automated annotation tools
Quantitative benchmarking across diverse biological contexts
Systematic evaluation of CellTypist against other automated annotation tools reveals distinct performance patterns across accuracy, speed, and resolution metrics. In a comprehensive benchmarking analysis across six scRNA-seq datasets from human and mouse tissues, CellTypist demonstrated particular strengths in processing speed while maintaining high annotation accuracy [79].
Table 1: Performance metrics of CellTypist compared to other automated annotation tools
| Tool | Accuracy Range | Speed | Key Strengths | Limitations |
|---|---|---|---|---|
| CellTypist | >94% across 5/6 datasets [79] | ~30x faster than scSorter [79] | Fast processing; granular immune cell subtypes; multi-tissue optimization [2] [79] | Performance depends on reference data comprehensiveness [2] |
| ScType | 98.6% (72/73 cell types) [79] | Ultra-fast [79] | Comprehensive marker database; negative marker incorporation; cancer cell identification [79] | Limited for novel cell types without database markers |
| scSorter | High accuracy (slightly below ScType) [79] | 30x slower than ScType [79] | Robust to technical noise; good for closely-related subtypes [79] | Slower processing speed |
| SingleR | High concordance with manual annotation [80] | Fast [80] | Excellent for spatial transcriptomics; easy implementation [80] | Reference dataset dependency |
| LLM-based (Claude 3.5 Sonnet) | 80-90% for major cell types [81] | Variable (API-dependent) [81] | De novo annotation capability; no reference required [81] | Inconsistency across model sizes; computational cost |
CellTypist achieves precision, recall, and global F1-scores of approximately 0.9 for cell type classification at both high- and low-hierarchy levels [2]. Its performance advantage is particularly evident in complex immune cell environments, where it successfully distinguishes between 43 specific immune subtypes, including CD4+ T cell helper, regulatory, and cytotoxic subsets, CD8+ T cell subpopulations, and three distinct dendritic cell subsets (DC1, DC2, and migDCs) [2]. This granular resolution makes CellTypist particularly valuable for comprehensive immune cell profiling across physiological and disease contexts.
Performance in spatial transcriptomics applications
With the rapid advancement of spatial transcriptomics technologies, benchmarking automated annotation tools in these contexts has become increasingly important. Recent evaluations of reference-based cell type annotation methods for 10x Xenium spatial transcriptomics data identified SingleR as the best-performing method, demonstrating high accuracy and speed with results closely matching manual annotation [80]. However, CellTypist's specialized training on immune cell types positions it as a valuable complementary approach for detailed immune cell characterization within spatial contexts, particularly when integrated with spatial information for validation.
Experimental protocols for benchmarking studies
Standardized workflow for performance evaluation
Implementing rigorous, reproducible benchmarking of automated cell type annotation tools requires standardized experimental protocols. The following workflow outlines a comprehensive approach for comparing CellTypist against alternative methods:
Figure 1: Workflow for benchmarking automated cell type annotation tools. The process begins with diverse dataset collection, progresses through standardized processing and annotation steps, and concludes with comprehensive evaluation metrics.
Data collection and preprocessing protocol
Dataset Selection: Curate diverse scRNA-seq datasets representing various tissues, species, and experimental conditions. For comprehensive immune cell benchmarking, include datasets with well-established immune cell diversity such as peripheral blood mononuclear cells (PBMCs), tumor microenvironments, and lymphoid tissues [79] [78].
Quality Control: Apply standardized filtering criteria to remove low-quality cells and genes using Scanpy or Seurat workflows. Typical thresholds include:
- Cells with fewer than 200 detected genes
- Genes detected in fewer than 3 cells
- Mitochondrial gene percentage below 20% [78]
Normalization: Normalize count matrices using the "LogNormalize" method in Seurat, scaling to 10,000 reads per cell and log-transforming the results [78]. Alternatively, use Scanpy's
sc.pp.normalize_totalandsc.pp.log1pfunctions for consistent processing.
Tool execution and evaluation protocol
CellTypist Implementation:
Comparative Tool Execution: Implement alternative methods using standardized parameters:
Evaluation Metrics: Calculate performance measures including:
- Accuracy: Percentage of correctly annotated cells compared to manual expert annotation
- F1-score: Harmonic mean of precision and recall
- Computational efficiency: Processing time and memory usage
- Resolution capability: Ability to distinguish closely related cell subtypes [79]
Table 2: Essential research reagents and computational tools for benchmarking studies
| Category | Resource | Specification | Application in Benchmarking |
|---|---|---|---|
| Reference Datasets | Tabula Sapiens v2 | Multi-tissue scRNA-seq atlas | Ground truth for annotation accuracy [81] |
| Human Cell Atlas | Comprehensive tissue collection | Reference for rare cell type identification [79] | |
| Marker Databases | ScType Database | >2000 cell-type specific markers | Standardized marker gene sets [79] |
| CellTypist Models | ImmuneAllLow.pkl, ImmuneAllHigh.pkl | Pre-trained classifiers for immune cells [1] | |
| Software Tools | Scanpy | v1.7.0+ | Data preprocessing and visualization [7] |
| Seurat | v4.3.0+ | Data integration and clustering [78] | |
| AnnDictionary | LLM-integrated package | Alternative annotation approach [81] | |
| Benchmarking Platforms | SPATCH Portal | Spatial transcriptomics data | Platform-specific performance evaluation [82] |
CellTypist model selection guidelines
CellTypist offers multiple pre-trained models optimized for different applications. Selection should be based on specific research needs:
- ImmuneAllLow.pkl: Default model providing balanced resolution across major immune cell types with faster processing [1]
- ImmuneAllHigh.pkl: High-resolution model capturing fine-grained immune cell subtypes at increased computational cost [1]
- Custom Models: For specialized applications, researchers can train custom models using their own annotated datasets with the CellTypist training pipeline [2]
Advanced applications and integration strategies
Multi-tissue immune cell analysis
CellTypist demonstrates particular strength in cross-tissue immune cell analysis, enabling consistent annotation across diverse biological contexts. The tool was originally developed and validated on a dataset of 357,211 high-quality cells from 16 different tissues from 12 organ donors, establishing its capability to resolve tissue-specific immune features while maintaining consistent classification metrics across tissues [2]. This cross-tissue performance represents a significant advantage over methods trained on limited tissue contexts.
Figure 2: Multi-tissue immune cell analysis workflow with CellTypist. The tool enables consistent annotation across tissues while identifying tissue-specific signatures and clonal architecture.
Integration with spatial transcriptomics data
While sequencing-based spatial transcriptomics platforms like Visium HD and Stereo-seq provide whole transcriptome coverage, imaging-based platforms such as Xenium and CosMx offer single-cell resolution with targeted gene panels [82] [80]. CellTypist can be effectively integrated into spatial analysis pipelines through several approaches:
Direct Annotation: Apply CellTypist directly to spatial data when sufficient genes are detected, particularly for immune cell identification [80]
Integrated Analysis: Combine scRNA-seq references annotated with CellTypist with spatial data using integration tools like RCTD or Cell2location [80]
Validation Framework: Use spatial protein expression patterns from CODEX to validate CellTypist annotations in adjacent tissue sections [82]
Addressing annotation challenges in complex datasets
Automated annotation tools face specific challenges in complex biological contexts. CellTypist incorporates several features to address these challenges:
Probability Match Mode: Alternative to the default "best match" mode that assigns the "Unassigned" label when no cell type passes the probability threshold (default: 0.5), or multiple labels when several cell types exceed the threshold [1]
Majority Voting: Post-processing step that refines initial predictions by considering the local cellular neighborhood, significantly improving annotation consistency [1] [4]
Batch Effect Mitigation: Demonstrated robustness to technical variations between datasets, though performance is enhanced when query datasets are processed similarly to training data [2]
Comprehensive benchmarking establishes CellTypist as a high-performance tool for automated immune cell annotation, with particular strengths in processing speed, immune subtype resolution, and cross-tissue consistency. Based on systematic evaluations, the following implementation guidelines are recommended:
For general immune cell annotation projects, CellTypist provides an optimal balance of accuracy and computational efficiency, particularly when working with diverse tissue samples. In specialized contexts requiring identification of closely related immune cell subtypes, ScType offers complementary strengths with its comprehensive marker database. For spatial transcriptomics data, SingleR currently demonstrates superior performance, though CellTypist annotations can provide valuable immune-specific validation.
Future development directions for CellTypist include expansion of non-immune cell type coverage, enhanced integration with spatial analysis pipelines, and incorporation of multi-modal data inputs. As automated annotation continues to evolve, rigorous benchmarking using the protocols outlined herein will remain essential for validating new methodologies and ensuring biological insights derived from single-cell and spatial transcriptomics data.
This application note details a robust framework for validating single-cell RNA sequencing (scRNA-seq) immune cell annotations, specifically those generated by the CellTypist tool, through integration with proteomic and flow cytometry data. As single-cell transcriptomics becomes a mainstay in immunology and drug development, confirming computational predictions with orthogonal protein-level measurements is crucial for generating reliable, actionable biological insights. This protocol provides a standardized methodology for this multi-omic validation, leveraging the complementary strengths of each technology to build a more complete and confident understanding of immune cell populations [83] [84].
The core challenge addressed is the inherent discrepancy between a cell's transcriptome and its proteome. While tools like CellTypist offer powerful, automated annotation of scRNA-seq data into specific immune cell types using reference atlases, these predictions are based solely on RNA [17]. This protocol outlines how to correlate these RNA-based classifications with surface and intracellular protein expression data from flow cytometry (a high-throughput cytometric technique) and global protein profiling from proteomics, thereby strengthening the validity of the identified cell types and states [77] [85].
The CellTypist ecosystem provides a standardized vocabulary and computational model for annotating immune cells from scRNA-seq data, often referencing the Cell Ontology for consistent nomenclature [17]. However, the transition from computational prediction to biological validation requires a multi-faceted experimental approach. Flow cytometry offers a high-throughput, targeted method for quantifying the expression of key protein markers on individual cells, directly mirroring the cell type definitions often used in computational classification [77] [85]. Proteomics, particularly when applied to sorted cell populations, provides a broader, unbiased view of the protein landscape, confirming not only the presence of key markers but also the functional state of the cell [84].
This document is structured to guide researchers through a sequential process: from initial computational annotation with CellTypist to experimental design for validation, and finally to integrated data analysis. By following this application note, researchers can enhance the rigor of their immune profiling studies, a critical step for applications in biomarker discovery, patient stratification, and therapeutic target identification [25] [86] [83].
Detailed Protocols
Protocol 1: Automated Immune Cell Annotation with CellTypist
Principle: CellTypist automates the annotation of cell types from scRNA-seq data by leveraging a curated reference atlas of immune cells. This serves as the foundational hypothesis for subsequent protein-level validation [17].
Workflow Diagram: CellTypist Annotation Process
Step-by-Step Procedure
Data Input Preparation:
- Prepare your scRNA-seq count matrix in a format compatible with CellTypist (e.g., Scanpy object).
- Ensure gene identifiers are standardized.
Model Selection and Application:
- Load the appropriate pre-trained CellTypist model. The Pan Immune Immune Atlas v2 is recommended for comprehensive immune cell annotation [17].
- Run the CellTypist algorithm on your dataset. The tool will assign each cell a probabilistic score for different cell types.
Output and Interpretation:
- Extract the cell type labels and corresponding confidence scores.
- Quality Control: Filter out cells with low-confidence annotations or ambiguous probabilities before proceeding to validation. Establish a confidence threshold (e.g., probability > 0.8) for robust predictions.
Key Reagent Solutions
| Item | Function/Description | Source/Reference |
|---|---|---|
| CellTypist Software | Automated cell type annotation tool for scRNA-seq data. | [17] |
| Pan Immune Atlas v2 | A curated reference of immune cell transcriptomes used for annotation. | [17] |
| Cell Ontology (CL) ID | Standardized vocabulary for cell type names (e.g., CL:0000236 for B cells). | [17] |
Protocol 2: Validation by Flow Cytometry
Principle: This protocol uses flow cytometry to confirm the protein expression of key markers identified by CellTypist predictions on the same or a biologically matched sample [77] [85].
Workflow Diagram: Flow Cytometry Validation
Step-by-Step Procedure
Panel Design:
- Based on the CellTypist-predicted cell types, design a flow cytometry panel targeting lineage and state-specific protein markers (see Table 1).
- Example: If CellTypist identifies a
Tregpopulation, include antibodies against CD3, CD4, CD25, CD127, and FoxP3 for intracellular staining [85].
Sample Staining and Acquisition:
- Prepare a single-cell suspension from the tissue or cell source matched to the scRNA-seq sample.
- Stain cells with the optimized antibody panel, including viability dye to exclude dead cells.
- Acquire data on a flow cytometer, collecting a sufficient number of events for robust statistical analysis.
Automated Data Analysis and Correlation:
- Clustering and Phenotyping: Use an automated tool like CytoPheno to assign marker definitions and descriptive cell type names to cell clusters based on protein expression. This minimizes manual gating bias and standardizes the process [77].
- Correlation with CellTypist: Compare the frequencies of specific immune cell populations (e.g., % CD4+ T cells, % memory B cells) obtained from flow cytometry (CytoPheno) with the proportions predicted by CellTypist from scRNA-seq data. A strong positive correlation (e.g., R² > 0.8) validates the computational annotations.
Key Reagent Solutions
| Item | Function/Description | Source/Reference |
|---|---|---|
| Fluorochrome-conjugated Antibodies | Target-specific proteins (CD markers) for cell population identification. | [85] |
| Viability Dye | Distinguish live cells from dead cells during analysis. | [85] |
| CytoPheno Software | Automated tool for assigning cell type names to post-clustered cytometry data. | [77] |
Protocol 3: Validation by Proteomic Analysis of Sorted Populations
Principle: This protocol combines the precision of cell sorting with the breadth of proteomics to validate CellTypist annotations at a global protein level and uncover functional insights [84].
Step-by-Step Procedure
Cell Sorting Based on CellTypist Predictions:
- Using flow cytometry, sort the cell populations of interest (e.g.,
Naive B cellsvs.Memory B cells) as predicted by CellTypist. - Collect a sufficient number of cells (typically >50,000) for downstream proteomic analysis.
- Using flow cytometry, sort the cell populations of interest (e.g.,
Sample Preparation for Proteomics:
- Lyse the sorted cell populations.
- Digest proteins using a protease like trypsin.
- Label peptides with isobaric tags (e.g., TMT) if performing multiplexed experiments.
Mass Spectrometry and Data Analysis:
- Analyze the peptides by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS).
- Identify and quantify proteins using database search engines.
- Validation Analysis:
- Confirm the presence of key marker proteins used in the original CellTypist annotation (e.g., confirm high MS4A1 (CD20) expression in sorted B cells).
- Perform differential expression analysis to identify proteins that are significantly enriched in one sorted population over another, confirming distinct functional states.
Data Integration and Analysis
The final and most critical phase is the integrated analysis of all three data modalities.
Diagram: Multi-Omic Integration Logic
Correlation Analysis
Create a summary table to quantitatively compare the results across the different platforms. This allows for a direct assessment of validation success.
Table 1: Example Multi-omic Validation Table for a PBMC Sample
| Cell Type (CellTypist Prediction) | scRNA-seq Frequency (%) | Flow Cytometry Frequency (%) | Key Marker RNA (CellTypist) | Key Marker Protein (Flow Cytometry) | Proteomic Validation (Sorted Populations) | Validation Outcome |
|---|---|---|---|---|---|---|
| Naive B cells | 8.5 | 9.1 | MS4A1, CD19, CD27- | CD19+, CD20+, CD27- | High expression of CD19, CD20 confirmed by MS | Strong Correlation |
| CD4+ T cells | 35.2 | 32.8 | CD3D, CD4 | CD3+, CD4+ | High expression of CD3E, CD4 confirmed by MS | Strong Correlation |
| Tregs | 2.1 | 1.8 | CD3D, CD4, FOXP3, IL2RA | CD3+, CD4+, FOXP3+, CD127lo | High expression of FOXP3, IL2RA confirmed by MS | Strong Correlation |
| Non-classical Monocytes | 4.5 | 5.2 | CD14, FCGR3A (CD16) | CD14+, CD16+ | High expression of CD14, FCGR3A confirmed by MS | Strong Correlation |
Advanced Integrated Analysis
For a deeper integration, tools like CellWalker2 can be employed. CellWalker2 uses a graph-based model to integrate multi-modal data (e.g., scRNA-seq and scATAC-seq) and can probabilistically map cell types and genomic features across different contexts and hierarchies [87]. This approach can be extended to incorporate protein-level data, creating a unified model of cell identity that incorporates transcriptomic, epigenomic, and proteomic evidence.
The Scientist's Toolkit
Table 2: Essential Research Reagents and Tools for Multi-omic Validation
| Category | Item | Function in Protocol |
|---|---|---|
| Computational Tools | CellTypist | Automated annotation of immune cell types from scRNA-seq data. [17] |
| CytoPheno | Automated phenotyping and naming of cell clusters from flow cytometry data. [77] | |
| CellWalker2 | Graph-based integration of multi-omic data for cell type mapping and comparison. [87] | |
| Wet-Lab Reagents | Antibody Panels (Flow Cytometry) | Targeted protein-level detection of cell surface and intracellular markers. [85] |
| Cell Sorting Reagents | Isolation of pure populations of predicted cell types for proteomics. | |
| Proteomics Kits (Lysis, Digestion, TMT) | Preparation of samples for global protein analysis by mass spectrometry. [84] | |
| Reference Databases | Cell Ontology (CL) | Provides standardized cell type nomenclature for consistent reporting. [17] |
| Protein Ontology (PRO) | Used by tools like CytoPheno for standardizing marker names. [77] |
The comprehensive characterization of age-related immune dynamics represents a formidable challenge in immunology, requiring the precise identification of numerous immune cell subsets across diverse donor populations. Single-cell RNA sequencing (scRNA-seq) has revolutionized our ability to dissect immune complexity, but the analysis of large-scale datasets—particularly those investigating age-related changes—demands robust, standardized annotation methods to ensure reproducible and accurate cell type identification [20]. The CellTypist automated annotation platform has emerged as an essential tool for this purpose, enabling researchers to consistently classify immune cells across extensive datasets and longitudinal studies [2].
This application note demonstrates how CellTypist was employed to validate key findings in a landmark 2025 aging immunity study published in Nature [20]. The research profiled peripheral immunity in more than 300 healthy adults (ages 25-90) using scRNA-seq, proteomics, and flow cytometry, following 96 adults longitudinally across two years with seasonal influenza vaccination. The resulting dataset of over 16 million peripheral blood mononuclear cells (PBMCs) required precise, consistent annotation across 71 immune cell subsets to identify age-related transcriptional reprogramming [20] [88]. Through this case study, we illustrate best practices for implementing CellTypist in aging immune research and provide detailed protocols for reproducing these analyses.
Experimental Background and Design
Study Objectives and Cohort Design
The primary objective of the original research was to understand how immune cell composition and states shift with age, chronic viral infection, and vaccination [20]. The experimental design incorporated two complementary cohorts:
- Longitudinal Cohort: 96 healthy adults (49 young: 25-35 years; 47 older: 55-65 years) followed over 2 years with annual influenza vaccination and 8-10 blood draws per donor
- Cross-sectional Validation Cohort: 234 healthy adults (40 to >90 years) for validation across advanced age ranges [20]
This design enabled both cross-age comparisons and longitudinal tracking of individual donors, capturing both homeostatic maintenance and vaccine-induced immune perturbations [19].
Multi-Omic Data Generation
The study generated three primary data types requiring integration:
- scRNA-seq: >16 million PBMCs using single-cell RNA sequencing
- Proteomics: Olink plasma proteomics measuring 69 differentially expressed proteins
- Flow Cytometry: Spectral flow cytometry for validation of cell composition [20]
Annotation Challenges in Aging Immune Studies
Aging immune studies present unique annotation challenges due to:
- Transcriptional reprogramming: T cells exhibit altered gene expression patterns with age that may blur conventional cell type boundaries [20]
- Compositional shifts: Changing frequencies of naive and memory subsets require consistent classification across age groups [89]
- Longitudinal consistency: Cell annotation must remain stable across multiple time points for accurate trajectory analysis [20]
Without standardized annotation, these factors introduce variability that can obscure genuine age-related signatures or generate false discoveries.
CellTypist Implementation for Age-Related Immune Dynamics
Model Selection and Configuration
For the aging immune study, researchers employed CellTypist with the following configuration:
- Model Selection: "ImmuneAllLow" (high-resolution) and "ImmuneAllHigh" (low-resolution) models
- Rationale: These models encompass immune cell types collected from different tissues, providing comprehensive coverage of peripheral blood immune subsets [14]
- Hierarchical Strategy: Applied a tiered approach with 9 cell subsets at level 1, 29 subsets at level 2, and 71 subsets at level 3 [19]
The "ImmuneAllLow" model was particularly valuable for resolving fine-grained T cell subsets that exhibited the most significant age-related transcriptional changes [20].
Annotation Workflow Integration
The CellTypist annotation process was integrated into the analytical pipeline as follows:
Figure 1: CellTypist Annotation Workflow. The workflow begins with quality-controlled scRNA-seq data, proceeds through model selection and automated annotation, and culminates in age correlation analyses.
Key Findings Enabled by CellTypist
Implementation of CellTypist enabled several critical discoveries in age-related immune dynamics:
- T Cell-Specific Reprogramming: Identified robust, non-linear transcriptional reprogramming in T cell subsets with age, not observed in other immune lineages [20]
- Early Differentiation Changes: Revealed that T cell subsets early in differentiation (naive T cells) showed the highest number of age-related differentially expressed genes [20]
- Stable Reprogramming Signature: Demonstrated that age-related transcriptional changes persisted over a 2-year longitudinal follow-up [20]
- TH2 Bias Discovery: Uncovered functional T helper 2 (TH2) cell bias in memory T cells linked to dysregulated B cell responses in older adults [20]
Table 1: Key Age-Related Immune Changes Identified Through CellTypist-Annotated scRNA-seq Data
| Immune Compartment | Key Age-Related Change | Functional Consequence | Citation |
|---|---|---|---|
| Naive CD4+ T Cells | 331 differentially expressed genes | Transcriptional reprogramming without frequency changes | [20] |
| Naive CD8+ T Cells | 182 differentially expressed genes | Both transcriptional and frequency alterations | [20] |
| Memory CD4+ T Cells | TH2 bias development | Dysregulated B cell responses to influenza vaccine | [20] |
| Circulating Proteome | 69 proteins differentially expressed | Increased CXCL17, WNT9A, GDF15 without classic inflammation | [20] |
Detailed Experimental Protocols
CellTypist Annotation Protocol for Aging Studies
Protocol 1: Automated Cell Annotation with CellTypist
Materials:
- Processed scRNA-seq data (post-quality control)
- CellTypist installation (Python package)
- ImmuneAllLow and ImmuneAllHigh models
Procedure:
- Data Preparation: Format scRNA-seq data as AnnData object with normalized counts
- Model Download:
- Cell Type Prediction:
- Hierarchical Annotation: Apply high-hierarchy model first, then refine with low-hierarchy model
- Quality Assessment: Validate annotations against known marker genes
- Cross-model Validation: Compare results across multiple models for consistency
Troubleshooting Tips:
- For low-heterogeneity datasets, implement majority voting to improve accuracy
- When annotating rare populations, consider increasing model sensitivity thresholds
- Validate problematic annotations with manual inspection of marker genes [22]
Age-Related Differential Expression Analysis Protocol
Protocol 2: Identifying Age-Associated Transcriptional Changes
Materials:
- CellTypist-annotated scRNA-seq data
- Differential expression framework (e.g., Scanpy, Seurat)
- Donor metadata including age, sex, CMV status
Procedure:
- Subset Identification: Isolate specific cell subsets using CellTypist annotations
- Pseudo-bulk Creation: Aggregate expression profiles within donors and cell types
- Differential Testing:
- Apply linear models accounting for age as continuous variable
- Include covariates (sex, CMV status) in model design
- Implement appropriate multiple testing correction
- Pathway Analysis: Conduct gene set enrichment on age-associated genes
- Trajectory Analysis: Project cells along putative aging trajectories using diffusion maps
Validation Steps:
- Confirm findings in independent validation cohort
- Correlate transcriptional changes with protein expression (proteomics)
- Validate key populations using flow cytometry [20]
Longitudinal Analysis Protocol
Protocol 3: Analyzing Temporal Immune Dynamics
Materials:
- Longitudinal scRNA-seq data with multiple time points
- CellTypist annotations consistent across time points
- Vaccination response metadata
Procedure:
- Time-point Alignment: Ensure consistent annotation across all collection time points
- Compositional Tracking: Monitor changes in cell type frequencies over time
- Stability Assessment: Calculate RNA Age Metric (RAM) for T cell subsets
- Vaccination Response Analysis: Compare pre- and post-vaccination cell states
- Cross-sectional Validation: Verify longitudinal findings in independent cohort
Key Metrics:
- RNA Age Metric (RAM): Composite score of age-related DEGs [20]
- IMM-AGE: Cellular frequency-based aging metric [20]
- Immune Health Metric (IHM): Whole-blood transcriptomics-based metric [20]
Table 2: Essential Research Resources for Age-Related Immune Dynamics Studies
| Resource Category | Specific Tool/Reagent | Application in Aging Studies | Validation in Study |
|---|---|---|---|
| Annotation Tools | CellTypist | Automated immune cell annotation | Annotated 71 immune subsets across 16M cells [2] |
| Reference Databases | Human Immune Health Atlas | Age-inclusive reference framework | Incorporated 108 donors (11-65 years) [19] |
| Experimental Models | ImmuneAllLow Model | High-resolution immune cell typing | Resolved 35 T cell subsets showing age effects [20] |
| Validation Tools | Spectral Flow Cytometry | Protein-level validation of annotations | Confirmed compositional changes with age [20] |
| Longitudinal Metrics | RNA Age Metric (RAM) | Quantifying transcriptional aging | Tracked T cell reprogramming stability [20] |
Data Integration and Visualization Framework
The aging immune dynamics study required sophisticated integration of multiple data types, all anchored by consistent CellTypist annotations:
Figure 2: Multi-omic Data Integration Framework. CellTypist annotations serve as the foundation for integrating transcriptomic, proteomic, and flow cytometry data to discover age-related immune patterns.
This application note demonstrates how CellTypist enables robust, reproducible annotation of scRNA-seq data in age-related immune dynamics studies. The implementation of CellTypist in the referenced Nature study provided several critical advantages:
- Consistency Across Age Groups: Automated annotation ensured equivalent cell type definitions across diverse age ranges
- Longitudinal Stability: Consistent classification enabled tracking of immune subsets across multiple time points
- High-Resolution Discovery: Fine-grained cell typing revealed subset-specific aging patterns
- Multi-study Integration: Standardized annotations facilitated comparison with external datasets
For researchers investigating immune aging, we recommend:
- Beginning with the "ImmuneAllLow" model for high-resolution analysis
- Implementing hierarchical validation strategies
- Correlating transcriptional findings with protein-level measurements
- Utilizing the provided public resources (https://apps.allenimmunology.org/aifi/insights/dynamics-imm-health-age/) for additional validation
The integration of CellTypist into the analytical pipeline proved essential for revealing the nuanced patterns of immune aging that would be challenging to detect with inconsistent manual annotations. This approach establishes a framework for future studies investigating immune dynamics across the human lifespan.
Within the framework of immune cell annotation research using CellTypist, assessing the reliability of automated cell type predictions is paramount for generating biologically meaningful results. CellTypist provides two primary quantitative metrics for this purpose: the decision score and the probability metric [90]. The decision matrix contains the raw decision scores for each cell across all cell types in the reference model, representing the distance of the cell from the classification hyperplane [90] [1]. The probability matrix, derived by applying the sigmoid function to the decision matrix, transforms these scores into a more interpretable range of 0 to 1, representing the probability each cell belongs to a given cell type [90] [1]. Understanding and properly utilizing these metrics enables researchers to distinguish between high-confidence annotations and ambiguous assignments, which is particularly crucial when dealing with novel cell states or closely related immune cell subtypes.
Core confidence metrics explained
Decision scores: The foundation of classification
The decision score forms the foundational output of CellTypist's logistic regression classifier. For each cell, a decision score is calculated for every cell type in the reference model, with higher scores indicating a stronger match to that particular cell type [90]. The cell type with the highest decision score is typically assigned as the predicted label when using the default 'best match' mode [1]. These scores are valuable for understanding the relative separation between potential cell type assignments for a given cell, as they represent the distance from the classification boundary in the high-dimensional feature space.
Probability metrics: Interpretable confidence measures
The probability matrix in CellTypist provides a normalized, more biologically interpretable measure of annotation confidence [90]. By applying the sigmoid function to the decision matrix, values are transformed to a 0-1 scale, where the probability represents the model's confidence that a cell belongs to a particular cell type [1]. These probabilities are particularly valuable for implementing multi-label classification, where cells can be assigned to zero, one, or multiple cell types based on a user-defined probability threshold [6] [1]. This approach is especially useful for identifying cells with ambiguous identities or capturing cells undergoing transitional states in immune processes.
Table 1: Key Differences Between Decision Scores and Probability Metrics in CellTypist
| Feature | Decision Score | Probability Metric |
|---|---|---|
| Origin | Raw output from logistic regression classifier | Transformation of decision score using sigmoid function |
| Range | Unbounded continuous values | Bounded between 0 and 1 |
| Interpretation | Distance from classification boundary | Probability of cell type membership |
| Primary Use | Understanding model mechanics and relative cell type similarities | Setting confidence thresholds and multi-label classification |
| Output File | decision_matrix.csv |
probability_matrix.csv |
Quantitative assessment of confidence metrics
The practical application of these confidence metrics can be quantitatively assessed across different immune cell types. Evaluation studies demonstrate that reliability varies significantly across cell types, with well-defined populations like B cells and T cells typically showing higher confidence scores compared to more heterogeneous or transitional populations such as dendritic cell subsets or proliferating cells [22]. The relationship between decision scores and probability metrics follows a sigmoidal curve, where moderate decision scores translate to probabilities near 0.5 with high uncertainty, while extreme decision scores produce probabilities approaching 0 or 1 with correspondingly high confidence [90] [1].
Table 2: Typical Confidence Ranges and Interpretations in Immune Cell Annotation
| Probability Range | Decision Score Relationship | Confidence Level | Recommended Action |
|---|---|---|---|
| 0.9 - 1.0 | Strongly positive | Very High | Confident assignment; suitable for definitive analysis |
| 0.7 - 0.9 | Moderately positive | High | Reliable assignment; minor uncertainty |
| 0.5 - 0.7 | Weakly positive | Moderate | Consider multi-label classification or manual inspection |
| 0.3 - 0.5 | Near classification boundary | Low | Likely ambiguous identity; requires additional validation |
| 0.0 - 0.3 | Weakly to strongly negative | Very Low | Cell type unlikely; potentially novel or poor quality cell |
Experimental protocol for confidence assessment
Workflow for comprehensive confidence evaluation
The following protocol details the complete workflow for assessing annotation confidence using decision scores and probability metrics in CellTypist, from data preparation through final interpretation. This workflow integrates both standard and advanced analytical approaches to provide researchers with a comprehensive framework for validating immune cell annotations.
Data preparation and model selection
Begin by preparing your single-cell RNA sequencing data as a raw count matrix with cells as rows and gene symbols as columns [1] [8]. The data should be in one of the accepted formats: .txt, .csv, .tsv, .tab, .mtx, or .mtx.gz [1]. For immune cell annotation, select an appropriate model based on your research context; the Immune_All_Low.pkl model is recommended as a starting point for high-resolution immune cell typing, while Immune_All_High.pkl provides broader immune categories [14] [8]. Models can be downloaded and inspected within Python:
Cell type prediction and confidence matrix generation
Execute cell typing using the celltypist.annotate function. For standard analysis, use mode = 'best match' to assign each cell to the single cell type with the highest score [1]. To generate both decision and probability matrices while enabling multi-label classification, implement the following:
Confidence threshold optimization and validation
Evaluate the distribution of probability scores across your dataset to identify an appropriate threshold for your specific biological context. The default threshold of 0.5 is suitable for most applications, but this can be adjusted based on the trade-off between assignment stringency and cell retention [1]. Cells with probabilities below your threshold for all cell types should be flagged as 'Unassigned' and may represent low-quality cells, doublets, or novel cell states not represented in the reference model [6]. For validation, compare the expression of canonical marker genes with the predicted cell types:
Advanced refinement using majority voting
For enhanced annotation accuracy, particularly in heterogeneous immune cell populations, implement majority voting refinement. This approach performs over-clustering of the query data and assigns the most frequent cell type label within each local cluster, effectively smoothing annotations across transcriptionally similar cells [8]. While this refinement improves consistency, it may increase computational time for large datasets due to the additional clustering step.
Table 3: Key Research Reagent Solutions for CellTypist-Based Immune Cell Annotation
| Resource | Function | Implementation Example |
|---|---|---|
| CellTypist Python Package | Core analytical toolbox for automated cell type annotation | pip install celltypist or conda install -c bioconda -c conda-forge celltypist [1] |
| Pre-trained Immune Cell Models | Reference classifiers for immune cell types | Immune_All_Low.pkl (high-resolution) or Immune_All_High.pkl (broad categories) [14] |
| scRNA-seq Data Format Converters | Prepare expression matrices in CellTypist-compatible formats | Scanpy (sc.read_10x_mtx, sc.read_csv) or Seurat (as.SingleCellExperiment) [1] |
| Marker Gene Validation Tools | Verify annotation confidence through expression checking | Scanpy plotting functions (sc.pl.dotplot, sc.pl.umap) [1] |
| Cell Ontology References | Standardize cell type terminology and definitions | Bionty CellType ontology (bt.CellType.public()) [17] |
Interpretation guidelines and troubleshooting
Decision score and probability relationships
The interpretation of confidence metrics requires understanding their relationship and limitations. High decision scores correspond to probabilities approaching 1, indicating confident assignments, while scores near zero produce probabilities near 0.5, reflecting uncertainty [90]. Systematic low confidence across many cells may indicate issues with data quality, incorrect model selection, or substantial batch effects. When consistently low probabilities are observed, consider the following troubleshooting approaches: (1) verify that your data is properly normalized using the same method as the training data (log1p normalized to 10,000 counts per cell) [8], (2) confirm that major cell populations in your data are represented in the reference model, and (3) check for potential batch effects that might distort the projection of your data into the reference model's feature space.
Advanced multi-label classification strategies
For complex immune datasets containing transitional states or cells with mixed identities, implement multi-label classification to capture this biological complexity. In this mode, set mode = 'prob match' and adjust p_thres based on your desired stringency [6] [1]. A conservative threshold (e.g., 0.7) will assign fewer multiple labels but with higher confidence, while a more liberal threshold (e.g., 0.3) will capture more ambiguous cases but require additional validation. Cells receiving multiple labels can represent biologically meaningful intermediate states, such as neutrophil-to-macrophage transitions or activated T cell phenotypes, which are particularly relevant in immune response studies.
Visualization and reporting of confidence metrics
Effective visualization of confidence metrics enhances the interpretability and communication of your results. Generate UMAP plots colored by confidence scores to identify patterns in prediction uncertainty across the cellular landscape [1]. Additionally, create violin plots showing the distribution of confidence scores by cell type to identify systematically problematic annotations. For publication-quality reporting, export the complete results:
The decision scores and probability metrics provided by CellTypist form a robust framework for assessing annotation confidence in immune cell profiling studies. By systematically applying these metrics through the protocols outlined above, researchers can significantly enhance the reliability of their single-cell RNA sequencing analyses, leading to more accurate biological interpretations and downstream applications in immunology and drug development.
Cross-validation Techniques for Custom Model Development
Cross-validation represents a fundamental statistical methodology in machine learning model development, particularly crucial for ensuring the reliability and generalizability of custom CellTypist models in immune cell annotation research. Within the broader thesis context of utilizing CellTypist for immune cell annotation, rigorous validation protocols ensure that models accurately capture immune cell heterogeneity rather than overfitting to technical artifacts or dataset-specific biases. CellTypist employs logistic regression classifiers optimized by stochastic gradient descent algorithms for cell type annotation [15]. When researchers develop custom models beyond the pre-built immune cell references, implementing robust cross-validation techniques becomes paramount for evaluating model performance and ensuring biological relevance.
The complexity of immune cell systems—with their continuous differentiation states, overlapping marker expressions, and context-dependent functional programs—demands validation approaches that account for multiple biological and technical variance sources. Cross-validation in this context serves not merely as a technical exercise but as a critical biological validation step ensuring that identified immune cell subsets correspond to genuine biological entities rather than analytical artifacts. This protocol details comprehensive cross-validation methodologies tailored specifically for CellTypist custom model development, with particular emphasis on immune cell annotation challenges and solutions.
Cross-Validation Framework for Immune Cell Annotation
Core Validation Concepts and Terminology
In CellTypist model development, several key concepts frame the cross-validation approach. Generalizability refers to a model's performance on unseen data, critically important for immune cell annotation where cellular states may vary across experimental conditions, tissues, and disease contexts. Overfitting occurs when models learn dataset-specific noise rather than biologically meaningful signal, a particular risk with high-dimensional scRNA-seq data capturing numerous immune cell states. Stratification ensures that rare immune cell populations are adequately represented across training and validation splits, preserving the prevalence of biologically important but computationally challenging minority subsets like tissue-resident memory T cells or dendritic cell subtypes.
The bias-variance tradeoff manifests distinctly in immune cell annotation, where overly simple models may collapse biologically distinct but transcriptionally similar populations (e.g., CD4+ T helper subsets), while overly complex models may fracture continuous activation gradients into artifactual discrete subtypes. Cross-validation techniques help navigate this tradeoff by providing empirical performance estimates across different model complexities. Performance metrics must be carefully selected for immune cell annotation tasks, with particular attention to metrics that appropriately weight rare populations and account for hierarchical relationships between cell types.
Cross-Validation Techniques Comparison
Table 1: Cross-Validation Techniques for CellTypist Custom Models
| Technique | Implementation Approach | Optimal Use Case | Advantages | Limitations |
|---|---|---|---|---|
| k-Fold Cross-Validation | Random partitioning into k equal subsets; k-1 folds for training, 1 fold for testing | Initial model development with balanced immune cell classes | Simple implementation, full dataset utilization | Potential data leakage, may not reflect real-world performance |
| Stratified k-Fold | Preservation of immune cell type proportions in each fold | Datasets with rare immune cell populations | Maintains minority class representation, more reliable performance estimates | Complex implementation, computationally intensive |
| Leave-One-Out Cross-Validation (LOOCV) | Single cell as test set, all others as training set | Very small datasets (<1000 cells) | Minimal bias, maximum training data | High computational cost, high variance in performance estimates |
| Leave-One-Group-Out Cross-Validation | All cells from single donor/experiment as test set | Multi-donor or multi-experiment datasets | Measures cross-donor generalizability, identifies batch effects | Requires multiple independent sample sources |
| Nested Cross-Validation | Outer loop for performance estimation, inner loop for hyperparameter tuning | Hyperparameter optimization and unbiased performance evaluation | Unbiased performance estimates, robust hyperparameter selection | Extremely computationally intensive |
| Hierarchical Cross-Validation | Account for dataset structure (cells within samples within donors) | Complex multi-level experimental designs | Appropriate for hierarchical data structure, prevents data leakage | Implementation complexity, requires careful experimental design documentation |
Experimental Protocols for Cross-Validation
Standard k-Fold Cross-Validation Implementation
The following protocol details the implementation of k-fold cross-validation for custom CellTypist models, with specific adaptations for immune cell annotation tasks:
Materials and Reagents:
- Processed single-cell RNA sequencing data in AnnData format
- CellTypist installation (version 1.0 or higher)
- Python environment (3.8 or higher) with scikit-learn, pandas, and numpy
- Pre-annotated reference dataset with immune cell labels
- Computational resources (minimum 16GB RAM for typical datasets)
Procedure:
- Data Preparation: Load and preprocess single-cell data using standard CellTypist pipeline, including quality control, normalization, and feature selection. Preserve immune cell heterogeneity by avoiding over-aggressive filtering.
Stratified Fold Generation: Implement stratified k-fold splitting to maintain immune cell subtype proportions:
Iterative Training and Validation: For each fold, train custom CellTypist model and validate performance:
Performance Aggregation: Calculate mean and standard deviation of performance metrics across all folds to assess model consistency and generalizability.
Advanced Cross-Validation for Complex Immune Datasets
For datasets with complex experimental designs (multiple donors, conditions, or timepoints), implement leave-one-group-out cross-validation to accurately assess model generalizability across biological replicates:
Procedure:
- Group Identification: Identify the grouping variable (donor, experiment, or condition) that represents independent biological replicates.
- Group-Wise Splitting: Iteratively designate all cells from one group as test set, with remaining groups as training set.
- Batch Effect Assessment: Monitor performance differences between within-group and cross-group predictions to quantify batch effects.
- Model Adjustment: If significant performance degradation occurs in cross-group validation, implement batch correction techniques or include more diverse training data.
Performance Metrics and Interpretation
Key Metrics for Immune Cell Annotation
Table 2: Performance Metrics for Immune Cell Annotation Models
| Metric | Calculation | Interpretation | Application Context |
|---|---|---|---|
| Overall Accuracy | Correct predictions / Total predictions | General model performance | Balanced datasets with uniform cell type distribution |
| Balanced Accuracy | Average of per-class accuracy | Performance on imbalanced datasets | Datasets with rare immune cell populations |
| F1 Score | Harmonic mean of precision and recall | Balance between false positives and false negatives | Critical applications where both false positives and negatives carry cost |
| Macro F1 | Unweighted mean of class-specific F1 scores | Performance across all classes regardless of prevalence | Comprehensive assessment of multi-class performance |
| Weighted F1 | Prevalence-weighted mean of F1 scores | Performance weighted by class importance | Contexts where majority classes are more important |
| Hierarchical F1 | F1 score accounting for cell type ontology | Performance at appropriate annotation granularity | When cell type relationships form hierarchy (e.g., T cells → CD4+ T cells → Th1 cells) |
| Cross-Donor Consistency | Performance variation across donors | Model robustness to biological variation | Multi-donor datasets assessing generalizability |
Interpreting Cross-Validation Results
Effective interpretation of cross-validation results requires understanding both statistical and biological implications. High variance in performance across folds suggests model instability or dataset heterogeneity that may require additional regularization or dataset expansion. Consistent misannotation patterns across folds indicate fundamental challenges in distinguishing specific immune cell subsets, potentially requiring additional marker genes or alternative modeling approaches.
Performance discrepancies between cross-validation strategies provide valuable biological insights. Significant performance degradation in leave-one-donor-out versus standard k-fold validation suggests donor-specific batch effects or biological variation that limits model generalizability. In such cases, incorporating more diverse donor samples or implementing explicit batch correction in the model training process becomes necessary.
Integration with CellTypist Workflow
Custom Model Training with Cross-Validation
The cross-validation process integrates seamlessly with CellTypist's custom model training pipeline, providing empirical evidence for model selection and optimization decisions. After identifying the optimal model configuration through cross-validation, researchers can train the final model on the complete dataset for maximum predictive power:
Majority Voting Enhancement
CellTypist's majority voting approach, which refines predictions through over-clustering and consensus labeling, can be validated through cross-validation to ensure it improves rather than degrades performance [41]. Implement cross-validation both with and without majority voting to quantify its contribution to annotation accuracy, particularly for heterogeneous immune cell populations:
Visualizing Cross-Validation Workflows
Figure 1: Cross-Validation Workflow for CellTypist Model Development. This diagram illustrates the iterative process of model training and validation across data partitions, culminating in model selection based on aggregated performance metrics.
Figure 2: Hierarchical Organization of Immune Cell Types. This hierarchical structure informs stratified cross-validation approaches and hierarchical performance metrics that account for annotation granularity.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Reagent/Tool | Specification | Function in Workflow | Implementation Notes |
|---|---|---|---|
| CellTypist Software | Python package v1.0+ | Core classification engine | Install via pip: pip install celltypist |
| Single-Cell Data | Processed AnnData objects (.h5ad) | Model input format | Requires quality control, normalization, and batch correction |
| Reference Annotations | Cell type labels with ontology | Ground truth for training | Should include hierarchical relationships where possible |
| scikit-learn | Python ML library | Cross-validation implementation | Provides KFold, StratifiedKFold, LeaveOneGroupOut implementations |
| Scanpy | Python single-cell analysis toolkit | Data preprocessing and visualization | Enables quality control, clustering, and visualization |
| CELLxGENE Census | Curated single-cell data | Reference data for model benchmarking | Provides standardized annotations across multiple datasets |
| CellHint | Cell type purity tool | Reference dataset purification | Ensures annotation accuracy and cell subtype purity [91] |
Cross-validation techniques provide an essential methodological foundation for developing robust, generalizable custom CellTypist models in immune cell annotation research. By implementing stratified, group-wise, and hierarchical validation approaches appropriate to the biological complexity of immune systems, researchers can confidently deploy models that capture genuine biological signals rather than dataset-specific artifacts. The integration of these validation techniques throughout the model development lifecycle—from initial feature selection through final performance assessment—ensures that resulting models will maintain predictive accuracy when applied to new datasets, experimental conditions, and biological contexts. As single-cell technologies continue to evolve and immune cell atlases expand, these cross-validation methodologies will remain critical for translating computational models into biologically meaningful insights with therapeutic relevance.
Addressing Tissue-Specific and Species-Specific Annotation Challenges
Automated cell type annotation using tools like CellTypist has significantly advanced the analysis of single-cell RNA sequencing (scRNA-seq) data. However, researchers consistently face two fundamental challenges that impact annotation accuracy: tissue-specific heterogeneity and species-specific differences. Immune cells, central to numerous physiological and pathological processes, exhibit remarkable functional and phenotypic plasticity across different tissue environments and between model organisms and humans. Addressing these challenges is critical for ensuring the biological validity of single-cell analyses in immunology research and drug development. This Application Note provides detailed protocols and frameworks for using CellTypist to overcome these specific annotation hurdles, ensuring reliable and reproducible immune cell identification across diverse research contexts.
Quantitative Challenges in Tissue and Species Annotation
Performance Variation Across Tissue Types
Recent large-scale validation studies reveal significant performance variation in automated annotation tools across tissues with different cellular heterogeneity. The following table summarizes annotation consistency rates for various tools across diverse biological contexts:
Table 1: Annotation Performance Across Tissue Types and Methods
| Tool/Method | PBMCs (High Heterogeneity) | Gastric Cancer | Embryo (Low Heterogeneity) | Stromal Cells (Low Heterogeneity) | Species-Specific Capabilities |
|---|---|---|---|---|---|
| LICT (LLM-based) | 90.3% match rate [22] | 91.7% match rate [22] | 48.5% match rate [22] | 43.8% match rate [22] | Not specified |
| CellTypist (ImmuneAllLow) | 65.4% exact match [31] | Not specified | Not specified | Not specified | Human and mouse models [14] |
| CellTypist (ImmuneAllHigh) | Higher-level annotations [14] | Not specified | Not specified | Not specified | Human and mouse models [14] |
| STCAT (T cell focus) | Not specified | 28% higher accuracy than existing tools [25] | Not specified | Not specified | Human-specific [25] |
The data demonstrates that low-heterogeneity tissues (embryonic and stromal cells) present particular challenges, with even advanced methods achieving less than 50% annotation consistency compared to manual curation [22]. This has direct implications for immune cell annotation in non-lymphoid tissues where immune populations may exhibit transitional states or tissue-specific adaptations.
Impact of Cellular Heterogeneity on Annotation Reliability
The reliability of automated annotation is directly influenced by the degree of cellular heterogeneity within samples:
Table 2: Impact of Cellular Heterogeneity on Annotation Tools
| Heterogeneity Level | Example Tissues | Primary Challenges | CellTypist Strategy |
|---|---|---|---|
| High Heterogeneity | PBMCs, Spleen, Lymph Nodes | Distinguishing closely-related immune subsets | Use low-hierarchy models (e.g., Immune_All_Low.pkl) [14] |
| Low Heterogeneity | Stromal cells, Embryonic tissues, Brain regions | Limited discriminatory marker genes | Use high-hierarchy models initially, then refine [14] |
| Intermediate Heterogeneity | Solid Tumors, Inflamed Tissues | Mixed cell states, activation continuum | Combine majority voting with manual validation [1] |
Performance disparities across tissue types underscore the importance of selecting appropriate CellTypist models and implementing validation strategies tailored to the specific biological context of your research.
Experimental Protocols for Challenging Contexts
Protocol 1: Cross-Tissue Immune Cell Annotation
Purpose: To accurately annotate immune cells across tissues with different heterogeneity profiles using CellTypist's model hierarchy.
Materials:
- CellTypist installation (Python 3.6+) [1]
- Query dataset in appropriate format (
.txt,.csv,.tsv,.tab,.mtx, or.mtx.gz) [1] - CellTypist models:
Immune_All_High.pklandImmune_All_Low.pkl[14]
Methodology:
- Data Preprocessing:
- Prepare your single-cell count matrix with cells as rows and genes as columns
- Ensure gene symbols match those used in CellTypist reference models
- Include non-expressed genes in the input table as they provide negative transcriptomic signatures [1]
Hierarchical Annotation Strategy:
- Begin with high-hierarchy annotation using
Immune_All_High.pkl: - Progress to low-hierarchy annotation using
Immune_All_Low.pklfor refined subset identification:
- Begin with high-hierarchy annotation using
Majority Voting Implementation:
- Apply majority voting to consolidate cell-type predictions:
Validation and Quality Control:
- Export probability matrices to assess prediction confidence:
- Compare high and low-hierarchy results to identify discordant annotations requiring manual inspection
Troubleshooting:
- For tissues with unusual immune populations (e.g., immune-privileged sites), use the probability match mode (
mode = 'prob match') to identify cells that fail probability thresholds [1] - When annotations seem biologically implausible, validate against known marker genes from external resources like CellMarker or PanglaoDB [92]
Protocol 2: Species-Specific Adaptation Strategy
Purpose: To adapt CellTypist for annotating immune cells across species despite primary training on human data.
Materials:
- CellTypist installation with all available models [1]
- Species-specific marker gene database (e.g., PanglaoDB) [92]
- Orthology mapping resource (e.g., Ensembl Compara)
Methodology:
- Reference Model Selection:
- Identify the most appropriate pre-trained model based on your target species:
- For mouse immunology studies, utilize specifically trained mouse immune models
Cross-Species Gene Symbol Mapping:
- Map gene symbols between species using orthology databases
- Create a converted count matrix with human ortholog symbols for analysis with human-trained models
Custom Model Development (for non-human species):
- Train a custom CellTypist model when pre-trained models are insufficient:
- Utilize species-specific reference atlases when available (e.g., mouse cell atlas)
Validation of Cross-Species Predictions:
- Implement multi-label classification to identify cells with ambiguous species-specific identities:
- Manually validate unexpected annotations using species-specific marker genes
Interpretation Guidelines:
- Exercise caution when interpreting annotations for cell types with known species-specific differences (e.g., NK cell subsets, γδ T cells)
- For evolutionary comparisons, focus on conserved immune populations (macrophages, conventional T cells) rather than species-specific subsets
Visual Workflow for Addressing Annotation Challenges
Workflow for Tissue-Specific Annotation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Addressing Annotation Challenges
| Resource | Type | Primary Function | Application Context |
|---|---|---|---|
CellTypist Immune_All_Low.pkl |
Pre-trained model | High-resolution immune cell annotation | Identifying closely-related immune subsets in high-heterogeneity tissues [14] |
CellTypist Immune_All_High.pkl |
Pre-trained model | Broad immune cell categorization | Initial annotation of low-heterogeneity tissues or novel immune populations [14] |
| PanglaoDB | Marker gene database | Species-specific marker gene reference | Validating automated annotations across human and mouse datasets [92] |
| CellMarker | Marker gene database | Curated cell type markers | Resolving ambiguous annotations in tissue-specific contexts [31] |
| AIDA (Asian Immune Diversity Atlas) | Reference dataset | Benchmarking annotation performance | Evaluating cross-population annotation accuracy [31] |
| TCellAtlas | Specialized database | T cell subtype reference | Annotating complex T cell states in disease contexts [25] |
Advanced Integration Strategies
Multi-Model Integration for Challenging Tissues
For particularly difficult annotation scenarios involving low-heterogeneity tissues or novel cellular states, consider implementing a multi-model integration approach similar to that used in LICT [22]. This strategy leverages the complementary strengths of multiple annotation methods:
Parallel Annotation Pipeline:
- Process query data through both CellTypist and supplementary tools (e.g., scGPT, SingleR)
- Identify consensus annotations across multiple methods
- Flag discordant predictions for special attention
Iterative Validation Protocol:
- Use the "talk-to-machine" approach to refine ambiguous annotations [22]
- Incorporate marker gene validation directly into the annotation workflow:
Objective Credibility Assessment
Implement an objective credibility evaluation framework to distinguish technical artifacts from biologically meaningful annotations [22]:
Marker Gene Expression Thresholding:
- For each predicted cell type, verify expression of canonical markers
- Establish minimum expression thresholds (e.g., >4 marker genes expressed in ≥80% of cells) [22]
- Flag populations failing these criteria for re-examination
Contextual Plausibility Assessment:
- Evaluate whether annotated cell types are biologically plausible in the tissue context
- Cross-reference with expected immune composition based on literature
- Utilize domain expertise to resolve computationally-generated anomalies
Addressing tissue-specific and species-specific challenges in immune cell annotation requires a sophisticated approach that combines appropriate tool selection, rigorous validation, and biological expertise. CellTypist provides a robust foundation for automated annotation, but optimal performance across diverse experimental contexts depends on implementing the hierarchical strategies, validation protocols, and integration frameworks outlined in this Application Note. By adopting these practices, researchers can enhance the reliability of their immune cell annotations, leading to more biologically meaningful conclusions in immunology research and drug development.
CellTypist employs a logistic regression model, optimized via stochastic gradient descent (SGD), to automate the annotation of single-cell RNA sequencing (scRNA-seq) data [2] [15]. Its performance is quantitatively assessed using the metrics of precision, recall, and the global F1-score [2]. On a large-scale cross-tissue dataset encompassing nearly 360,000 immune cells, the CellTypist model demonstrated high performance, with precision, recall, and a global F1-score each reaching approximately 0.9 for cell type classification [2]. This indicates a strong balance between minimizing false positives and false negatives in cell type assignment.
Table 1: Key Performance Metrics for CellTypist
| Metric | Score | Interpretation |
|---|---|---|
| Precision | ~0.9 | High proportion of correctly identified cells among those predicted as a specific type |
| Recall | ~0.9 | High proportion of actual cell types that are successfully identified |
| Global F1-score | ~0.9 | Excellent harmonic mean of precision and recall |
Experimental Protocols for Accuracy Quantification
Model Training and Evaluation Protocol
The high accuracy of CellTypist is achieved through a rigorous multi-stage protocol.
Step 1: Reference Data Curation and Integration
- Objective: Assemble a comprehensive training dataset.
- Procedure: Integrate cells from 20 different tissues sourced from 19 deeply curated reference datasets [2]. Harmonize cell type annotations into a consistent hierarchy, encompassing both high-level (e.g., 32 types) and low-level (e.g., 91 subtypes) classifications [2].
Step 2: Classifier Training with Stochastic Gradient Descent
- Objective: Train a robust logistic regression classifier.
- Procedure: For large training datasets, implement SGD learning with mini-batch training [15]. Shuffle cells and bin them into equal-sized mini-batches (e.g., 1,000 cells per batch). Sequentially train the model using batches randomly sampled for a set number of epochs (typically 10-30) [15]. This process optimizes the model's coefficients for each gene and cell type.
Step 3: Model Performance Benchmarking
- Objective: Quantify model accuracy.
- Procedure: Apply the trained CellTypist model to a held-out query dataset. For each query cell, calculate a decision score for every cell type in the model's reference. The decision score is defined as the linear combination of the scaled gene expression and the model coefficients associated with a given cell type [15] [1].
- Calculation of Metrics:
- Precision: For each cell type, calculate the ratio of correctly predicted cells to all cells predicted as that type.
- Recall: For each cell type, calculate the ratio of correctly predicted cells to all cells truly belonging to that type.
- F1-score: Compute the harmonic mean of precision and recall for each cell type. The global F1-score is an aggregate measure across all cell types [2].
Cell Annotation Protocol with Majority Voting Refinement
This protocol details the application of a trained CellTypist model to a new query dataset, including steps to enhance annotation confidence.
Step 1: Data Preparation and Model Loading
- Input Data: Prepare a raw count matrix (cells-by-genes or genes-by-cells) in a supported format (e.g.,
.txt,.csv,.mtx) [1]. Ensure all genes, including non-expressed ones, are included as they contribute to negative transcriptomic signatures. - Model Selection: Download and load an appropriate pre-trained model, such as
Immune_All_Low.pklfor broad immune cell classification [1].
- Input Data: Prepare a raw count matrix (cells-by-genes or genes-by-cells) in a supported format (e.g.,
Step 2: Initial Cell Prediction
- Procedure: Use the
celltypist.annotate()function to assign initial labels. Each cell is assigned to the cell type with the highest decision score ("best match" mode) or, alternatively, can be assigned to multiple types or remain "Unassigned" based on a probability threshold ("prob match" mode) [1].
- Procedure: Use the
Step 3: Majority Voting to Refine Predictions
- Objective: Improve annotation consistency by leveraging transcriptional similarity between neighboring cells.
- Procedure: Enable the
majority_voting = Trueparameter during annotation. This executes the following sub-steps:- Over-clustering: The query dataset is over-clustered using the Leiden algorithm to generate many small, transcriptionally homogenous subclusters [15] [1].
- Vote Tallying: Within each subcluster, the predicted labels of all individual cells are collected.
- Label Re-assignment: Each subcluster is assigned the identity of the dominant cell type present among its constituent cells [15] [1]. This process helps correct for outlier predictions and reinforces consistent labels within biologically meaningful clusters.
Diagram 1: Majority voting workflow for robust annotation.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for CellTypist Analysis
| Item | Function |
|---|---|
| scRNA-seq Dataset | A raw UMI count matrix from a healthy or diseased tissue sample; serves as the fundamental input for cell type annotation [1]. |
| CellTypist Python Package | The core software tool that performs automated cell type annotation and majority voting analysis [1] [4]. |
| Pre-trained Classification Models | Curated reference models (e.g., Immune_All_Low.pkl) containing the gene coefficients and parameters needed for cell type prediction [1]. |
| Cell Type Encyclopedia | A knowledge base providing detailed biological information and marker genes for the cell types defined in the models; aids in result interpretation [4]. |
Comparative Context and Technical Architecture
While CellTypist achieves high accuracy with a logistic regression model, other deep learning approaches have been developed. For instance, scHDeepInsight uses a convolutional neural network (CNN) architecture that converts gene expression data into 2D images and employs a hierarchical classification loss, reporting an average accuracy of 93.2% [93]. Another tool, scKAN, leverages a Kolmogorov-Arnold network and claims a 6.63% improvement in macro F1-score over state-of-the-art methods, including CellTypist [94]. These comparisons highlight a trend toward more complex, non-linear models for challenging classification tasks.
The core of CellTypist's technology is a logistic regression classifier. The following diagram illustrates the architecture of this model and the flow of data from a single cell through the classification process.
Diagram 2: CellTypist logistic regression classifier architecture.
Reproducibility Best Practices Across Different Research Environments
Reproducibility is a foundational principle of the scientific method, ensuring that research findings are transparent, reliable, and trustworthy. In computational biology, reproducibility specifically refers to the ability to produce the same results using the same data, code, and analysis conditions, while replicability involves reaching similar conclusions using new data and independent methods [95]. The single-cell RNA sequencing (scRNA-seq) field, particularly immune cell annotation research, faces significant reproducibility challenges due to complex analytical pipelines and continuously evolving cell type definitions. This document establishes best practices for maintaining reproducibility when using CellTypist for immune cell annotation, providing a framework that balances analytical flexibility with scientific rigor.
CellTypist Framework and Reproducibility Advantages
CellTypist is a machine learning-based tool designed for automated cell type annotation of scRNA-seq data. Its framework incorporates several features that inherently enhance reproducibility compared to manual annotation approaches. The tool employs regularised logistic regression models with stochastic gradient descent learning, providing a consistent and mathematically defined approach to cell classification [4] [2]. Unlike subjective manual annotation, which relies on individual researcher interpretation of marker genes, CellTypist applies uniform decision boundaries across datasets, enabling direct comparison of results between laboratories and studies.
The CellTypist ecosystem includes curated reference models trained on immune cells from multiple human tissues, capturing a comprehensive spectrum of immune cell states and types [2]. This reference database is continually expanded and refined through community contributions, creating a living resource that reflects evolving biological knowledge while maintaining version control for reproducibility. The automated nature of CellTypist significantly reduces annotation time while eliminating intra- and inter-observer variability common in manual approaches [96].
Table 1: Key Reproducibility Features of CellTypist
| Feature | Reproducibility Benefit | Implementation |
|---|---|---|
| Pre-trained models | Standardized cell type definitions | Logistic regression with SGD |
| Model versioning | Traceable reference data | Numbered model releases |
| Majority voting | Consensus cell typing | Ensemble prediction approach |
| Command-line interface | Automated workflow integration | Python API and command line |
| Open-source code | Transparent methodology | GitHub repository |
Experimental Protocol: CellTypist for Immune Cell Annotation
Sample Preparation and Quality Control
Begin with high-quality scRNA-seq data from platform-specific processing pipelines (e.g., Cell Ranger for 10x Genomics data). Implement rigorous quality control measures including filtering by UMI counts, gene detection thresholds, and mitochondrial read percentage [97]. Document all QC parameters precisely, as these directly impact downstream annotation results.
Data Preprocessing and Integration
Process data using standard tools (Seurat/Scanpy) with documented normalization methods, variable feature selection, and batch correction approaches. CellTypist is robust to technical variations, but consistent preprocessing remains critical. For multi-sample studies, address batch effects using Harmony or similar methods before annotation [26].
CellTypist Annotation Workflow
The majority_voting=True parameter implements a consensus approach that refines initial predictions by considering the identities of neighboring cells in the dataset, enhancing annotation robustness [4]. For optimal reproducibility, specify the exact model version used (e.g., model = models.Model.load('CellTypist_Immune_All_Low.pkl')).
Validation and Interpretation
Validate CellTypist annotations through multiple approaches: (1) Examine expression of canonical marker genes for assigned cell types; (2) Perform differential expression analysis between clusters; (3) Compare with independent annotation methods (e.g., manual annotation based on key markers) [54]. This multi-pronged validation strategy ensures biological relevance beyond computational prediction.
Diagram Title: CellTypist Annotation Workflow
Reproducibility Framework and Documentation Standards
Computational Environment Management
Reproducibility requires precise capture of the computational environment. Use containerization platforms (Docker/Singularity) to encapsulate the complete analysis environment, including operating system, software versions, and dependencies. For package management, employ Conda environments with explicit version pinning, particularly for critical packages including CellTypist, Scanpy, and scikit-learn. Maintain a comprehensive software manifest documenting all tools with exact version numbers and accessibility information (URLs, repositories) [95].
Analysis Documentation and Metadata
Maintain detailed records of all analytical decisions through structured metadata files. The README document for any CellTypist-based project should include: data provenance, preprocessing parameters, CellTypist model version, majority voting implementation, and any post-processing steps. For complex analyses spanning multiple stages, consider developing a replication package similar to the approach used by Yale researchers documenting environment setup, file structures, and computational requirements [95].
Table 2: Essential Documentation Components for Reproducible CellTypist Analysis
| Documentation Component | Required Information | Example |
|---|---|---|
| Data Provenance | Original data source, processing pipeline | "10x Genomics, Cell Ranger 7.1.0" |
| Preprocessing | QC thresholds, normalization method | "mingenes=200, maxgenes=5000, mt_percent=10" |
| CellTypist Parameters | Model version, voting scheme | "ImmuneAllLow.pkl, majority_voting=True" |
| Computational Environment | Software versions, container images | "CellTypist 1.5.0, Python 3.9.18" |
| Validation Approach | Marker genes, independent methods | "CD4+ T cells: CD3D, CD4; Manual curation" |
Data and Code Sharing Practices
Public repositories should contain both raw data (when possible) and processed data, along with complete analysis scripts. For CellTypist analyses, this includes: the count matrix, CellTypist model file or specification, annotation script, and resulting cell type labels. Utilize version-controlled repositories (Git) with comprehensive commit messages tracking analytical decisions. When data cannot be shared publicly due to privacy concerns, provide detailed instructions for data access and synthetic datasets demonstrating the analytical approach [95] [98].
Reproducibility Validation and Quality Assurance
Technical Validation Measures
Implement multiple technical validation approaches to ensure CellTypist annotations are robust. Cross-dataset validation applies the same CellTypist model to multiple independent datasets addressing similar biological questions. Stability analysis examines how annotations change with parameter variations, such as different preprocessing thresholds or model confidence cutoffs. Comparison with orthogonal methods validates CellTypist predictions against protein markers (CITE-seq) or other annotation tools [96] [2].
Biological Plausibility Assessment
Beyond technical validation, assess biological plausibility through multiple approaches: (1) Examine whether annotated cell types match expected tissue composition; (2) Verify that annotated cells express appropriate marker genes; (3) Confirm that rare cell populations appear at biologically reasonable frequencies; (4) Check that activation states align with experimental conditions [26] [54]. This multi-faceted assessment ensures computational predictions reflect biological reality rather than technical artifacts.
Diagram Title: Multi-level Validation Strategy
Research Reagent Solutions for Reproducible scRNA-seq
Table 3: Essential Research Reagents and Computational Tools for Reproducible Immune Cell Annotation
| Reagent/Tool | Function | Reproducibility Consideration |
|---|---|---|
| 10x Genomics Chromium | Single-cell partitioning | Standardized chemistry across experiments |
| CellTypist | Automated cell annotation | Version-controlled models |
| Seurat/Scanpy | Data preprocessing | Documented parameters and versions |
| Harmony | Batch correction | Consistent application across datasets |
| CellBender/SoupX | Ambient RNA removal | Reduces technical variability |
| Conda/Docker | Environment control | Computational reproducibility |
| Git | Version control | Analytical transparency |
Reproducible research using CellTypist for immune cell annotation requires a comprehensive approach spanning experimental design, computational implementation, documentation, and validation. By adopting the practices outlined in this document—standardized workflows, detailed documentation, computational environment control, and multi-faceted validation—researchers can produce immune cell annotations that are both biologically insightful and scientifically robust. The integration of automated tools like CellTypist within a reproducibility-focused framework represents the future of rigorous single-cell immunology research, accelerating discoveries while maintaining scientific integrity.
Conclusion
CellTypist represents a powerful, scalable solution for automated immune cell annotation that balances computational efficiency with biological accuracy. By integrating foundational knowledge of cell type classification with practical methodological workflows, optimization strategies, and rigorous validation approaches, researchers can reliably annotate diverse immune cell populations across various biological contexts. The tool's continuous development, including recent GPU support and expanded model collections, positions it as an essential resource for advancing immunological research. Future directions include enhanced integration with multi-omic datasets, improved handling of transitional cell states, and applications in clinical settings for disease classification and biomarker discovery. As single-cell technologies continue to evolve, CellTypist's flexible framework and community-driven model development will play a crucial role in deciphering immune system complexity and accelerating therapeutic innovations.
References
- 1. Teichlab/celltypist: A tool for semi-automatic cell type ... [https://github.com/Teichlab/celltypist]
- 2. Cross-tissue immune cell analysis reveals tissue-specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7612735/]
- 3. Tutorials [https://www.celltypist.org/tutorials]
- 4. CellTypist | automated cell type annotation for scRNA-seq ... [https://www.celltypist.org/]
- 5. Assessing GPT-4 for cell type annotation in single-cell RNA ... [https://www.nature.com/articles/s41592-024-02235-4]
- 6. Using CellTypist for multi-label classification [https://celltypist.readthedocs.io/en/stable/notebook/celltypist_tutorial_ml.html]
- 7. Using CellTypist for cell type classification [https://celltypist.readthedocs.io/en/latest/notebook/celltypist_tutorial.html]
- 8. CellTypist | Guide for using the online interface [https://www.celltypist.org/tutorials/onlineguide]
- 9. Celltype prediction - GitHub Pages [https://nbisweden.github.io/workshop-scRNAseq/labs/scanpy/scanpy_06_celltyping.html]
- 10. Stochastic gradient descent [https://en.wikipedia.org/wiki/Stochastic_gradient_descent]
- 11. Welcome to CellTypist's documentation! — celltypist 1.7.1 ... [https://celltypist.readthedocs.io/]
- 12. Logistic Regression Using SGD from Scratch [https://satishkumarmoparthi.medium.com/logistic-regression-with-l2-regularization-using-sgd-from-scratch-893692b48362]
- 13. Training function — celltypist 1.7.1 documentation [https://celltypist.readthedocs.io/en/latest/celltypist.train.html]
- 14. Models [https://www.celltypist.org/models]
- 15. FAQ [https://www.celltypist.org/faq]
- 16. 1.5. Stochastic Gradient Descent [https://scikit-learn.org/stable/modules/sgd.html]
- 17. CellTypist [https://docs.lamin.ai/celltypist]
- 18. Data analysis methods [https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/analysis/]
- 19. Longitudinal Multi-omic Immune Profiling Reveals Age ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11419011/]
- 20. Multi-omic profiling reveals age-related immune dynamics ... [https://www.nature.com/articles/s41586-025-09686-5]
- 21. A Review of Single-Cell RNA-Seq Annotation, Integration, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10417635/]
- 22. Evaluation of cell type annotation reliability using a large ... [https://www.nature.com/articles/s42003-025-08745-x]
- 23. The Power of automated cell type annotation [https://singleron.bio/the-power-of-accurate-automated-cell-type-annotation/]
- 24. sc-ImmuCC: hierarchical annotation for immune cell types ... [https://pubmed.ncbi.nlm.nih.gov/37545533/]
- 25. An automatic annotation tool and reference database for T ... [https://www.sciencedirect.com/science/article/pii/S2095927325002889]
- 26. Reproducible single-cell annotation of programs ... [https://www.nature.com/articles/s41592-025-02793-1]
- 27. Advancing Automated Cell Type Annotation with Large ... [https://www.sciencedirect.com/science/article/pii/S2001037025004775]
- 28. Cross-tissue multicellular coordination and its rewiring in ... [https://www.nature.com/articles/s41586-025-09053-4]
- 29. Human Immune Health Atlas [https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/]
- 30. A pan-cancer comparative analysis of the cancer genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12332167/]
- 31. Get, set, annotate - an overview of cell type prediction ... [https://www.cellkb.com/blog/cell_type_annotation_review]
- 32. Cell type ontologies of the Human Cell Atlas [https://www.nature.com/articles/s41556-021-00787-7]
- 33. Automatic cell-type harmonization and integration across ... [https://www.sciencedirect.com/science/article/pii/S0092867423013120]
- 34. Benchmarking popV Ensemble Cell Type Annotations on ... [https://blog.latch.bio/p/benchmarking-popv-ensemble-cell-type]
- 35. Celltypist - bioconda [https://anaconda.org/bioconda/celltypist]
- 36. Prediction function — celltypist 1.7.1 documentation [https://celltypist.readthedocs.io/en/latest/celltypist.annotate.html]
- 37. Labeling your own data [https://apps.allenimmunology.org/aifi/resources/imm-health-atlas/labeling/]
- 38. Scaling cross-tissue single-cell annotation models - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10592700/]
- 39. Current annotation strategies for T cell phenotyping of ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1306169/full]
- 40. 使用CellTypist 进行免疫细胞类型分类 [https://cloud.tencent.com/developer/article/1926662]
- 41. 如何用CellTypist实现单细胞类型精准分类?2025最新半 ... [https://blog.csdn.net/gitblog_00620/article/details/153917842]
- 42. Enhanced interpretation of immune cell phenotype and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12143338/]
- 43. NatGenet | 细胞注释新工具,使用popV 在单细胞数据中进行 ... [https://cloud.tencent.com/developer/article/2479955]
- 44. scanpy.tl.umap [https://scanpy.readthedocs.io/en/stable/generated/scanpy.tl.umap.html]
- 45. scanpy.pl.embedding [https://scanpy.readthedocs.io/en/latest/api/generated/scanpy.pl.embedding.html]
- 46. Core plotting functions — scanpy - Read the Docs [https://scanpy.readthedocs.io/en/stable/tutorials/plotting/core.html]
- 47. Age-dependent immune profile in healthy individuals [https://pmc.ncbi.nlm.nih.gov/articles/PMC11520839/]
- 48. An integrated single-cell atlas of blood immune cells in aging [https://www.nature.com/articles/s41514-024-00185-x]
- 49. Single-cell atlas of healthy human blood unveils age ... [https://www.sciencedirect.com/science/article/pii/S1074761323004533]
- 50. PBMC fixation and processing for Chromium single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6050658/]
- 51. Single-cell RNA-sequencing of peripheral blood ... [https://www.nature.com/articles/s41467-022-30893-5]
- 52. Immune cell identifier and classifier (ImmunIC) for single cell transcriptomic readouts | Scientific Reports [https://www.nature.com/articles/s41598-023-39282-4]
- 53. Best practice in large-scale cross-dataset label transfer using ... [https://celltypist.readthedocs.io/en/latest/notebook/celltypist_tutorial_cv.html]
- 54. 11. Annotation — Single-cell best practices [https://www.sc-best-practices.org/cellular_structure/annotation.html]
- 55. A Hands-On Introduction to cuML for GPU-Accelerated ... [https://machinelearningmastery.com/a-hands-on-introduction-to-cuml-for-gpu-accelerated-machine-learning-workflows/]
- 56. How to GPU-Accelerate Model Training with CUDA-X Data ... [https://developer.nvidia.com/blog/how-to-gpu-accelerate-model-training-with-cuda-x-data-science/]
- 57. GPU-Accelerated Single-Cell RNA Analysis with RAPIDS- ... [https://developer.nvidia.com/blog/gpu-accelerated-single-cell-rna-analysis-with-rapids-singlecell/]
- 58. Accelerating Biology with GPUs [https://watershed.bio/resources/accelerating-biology-with-gpus]
- 59. ScaleSC: Supercharging Single Cell RNA-seq Analysis ... [https://bioinforx.com/web/blog.php?id=1000095]
- 60. CPU vs. GPU for Machine Learning [https://blog.purestorage.com/purely-technical/cpu-vs-gpu-for-machine-learning/]
- 61. Comparison of missing data handling methods for variant ... [https://pubmed.ncbi.nlm.nih.gov/41103673/]
- 62. A Two-Step Algorithm for Handling Block-Wise Missing ... [https://www.mdpi.com/2076-3417/15/7/3650]
- 63. Leveraging sequences missing from the human genome to ... [https://www.nature.com/articles/s43856-025-01067-3]
- 64. Optimization of clustering parameters for single-cell RNA ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2025.1562410/full]
- 65. Evaluation of cell type annotation reliability using a large ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462508/]
- 66. Consensus prediction of cell type labels in single-cell data ... [https://www.nature.com/articles/s41588-024-01993-3]
- 67. Multimodal hierarchical classification of CITE-seq data ... [https://www.sciencedirect.com/science/article/pii/S266723752400328X]
- 68. A cell atlas foundation model for scalable search of similar ... [https://www.nature.com/articles/s41586-024-08411-y]
- 69. What is Data Normalization? A Complete Guide for 2025 [https://improvado.io/blog/data-normalization]
- 70. How to Normalize Data (2025 Guide) - Knack [https://www.knack.com/blog/how-to-normalize-data/]
- 71. How to normalize data [https://newrelic.com/blog/best-practices/data-normalization]
- 72. Issues running with conda · Issue #92 · Teichlab/celltypist [https://github.com/Teichlab/celltypist/issues/92]
- 73. Failed to install with conda · Issue #2 · Teichlab/celltypist [https://github.com/Teichlab/celltypist/issues/2]
- 74. Leveraging the Cell Ontology to classify unseen cell types [https://www.nature.com/articles/s41467-021-25725-x]
- 75. The Cell Ontology 2016: enhanced content, modularization ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4932724/]
- 76. Biology-driven insights into the power of single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492631/]
- 77. Automated descriptive cell type naming in flow and mass ... [https://www.nature.com/articles/s41598-025-12153-w]
- 78. Immune cell type signature discovery and random forest ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1194745/full]
- 79. Fully-automated and ultra-fast cell-type identification using ... [https://www.nature.com/articles/s41467-022-28803-w]
- 80. Benchmarking cell type annotation methods for 10x Xenium ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06044-0]
- 81. Benchmarking cell type and gene set annotation by large ... [https://www.nature.com/articles/s41467-025-64511-x]
- 82. Systematic benchmarking of high-throughput subcellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12534522/]
- 83. 2025 Trends: Multiomics [https://www.genengnews.com/topics/omics/2025-trends-multiomics/]
- 84. Integrating Cytomics and Proteomics [https://www.sciencedirect.com/science/article/pii/S1535947620315000]
- 85. A Guide to Flow Cytometry Markers [https://www.bdbiosciences.com/en-sg/learn/science-thought-leadership/blogs/flow-cytometry-markers]
- 86. Multi-omic integration sets the path for early prevention ... [https://www.nature.com/articles/s41525-025-00491-7]
- 87. CellWalker2: Multi-omic discovery using hierarchical cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278637/]
- 88. Multi-omic profiling reveals age-related immune dynamics ... [https://pubmed.ncbi.nlm.nih.gov/41162704/]
- 89. an original study, systematic review and meta-analysis [https://immunityageing.biomedcentral.com/articles/10.1186/s12979-024-00480-x]
- 90. Prediction result — celltypist 1.7.1 documentation [https://celltypist.readthedocs.io/en/stable/celltypist.classifier.AnnotationResult.html]
- 91. 基于千万细胞图谱的泛血液细胞注释工具的开发与应用 [https://dissertation.imicams.ac.cn/docinfo.action?id1=28b0702074700d480adaf6f9d52311c5&id2=op5o%252Fg1%252FdFc%253D]
- 92. Annotating cell clusters in single cell RNA-seq datasets [https://pluto.bio/resources/Learning%20Series/annotating-clusters-in-scrnaseq]
- 93. scHDeepInsight: a hierarchical deep learning framework ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12502058/]
- 94. scKAN: interpretable single-cell analysis for cell-type-specific ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03779-0]
- 95. Reproducibility Matters: Yale Experts Lead Sessions on Research Integrity and Best Practices | Institution for Social and Policy Studies [https://isps.yale.edu/news/blog/2025/10/reproducibility-matters-yale-experts-lead-sessions-on-research-integrity-and-best]
- 96. Current annotation strategies for T cell phenotyping of single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10768068/]
- 97. Best Practices for Analysis of 10x Genomics Single Cell ... [https://www.10xgenomics.com/analysis-guides/best-practices-analysis-10x-single-cell-rnaseq-data]
- 98. Reproducibility and replicability in research: What 452 professors think in Universities across the USA and India - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11940819/]