How Single-Cell Foundation Models Work: A Guide for Biomedical Researchers
Single-cell foundation models (scFMs) are transformative AI tools trained on millions of single-cell datasets to decipher the complex language of cellular biology.
How Single-Cell Foundation Models Work: A Guide for Biomedical Researchers
Abstract
Single-cell foundation models (scFMs) are transformative AI tools trained on millions of single-cell datasets to decipher the complex language of cellular biology. This article provides a comprehensive overview for researchers and drug development professionals, detailing how these models leverage transformer architectures to analyze gene expression data for tasks like cell type annotation, perturbation prediction, and drug sensitivity analysis. We explore the core concepts, from tokenization of gene expression to self-supervised pretraining, and critically evaluate their real-world performance against traditional methods. The content also addresses current limitations, offers guidance for model selection and optimization, and discusses the future potential of scFMs in advancing personalized medicine and therapeutic discovery.
The New Paradigm: How Foundation Models are Decoding Cellular Language
Single-cell foundation models (scFMs) represent a revolutionary convergence of artificial intelligence and cellular biology, creating a new paradigm for understanding cellular linguistics. These models are defined as large-scale deep learning models pretrained on vast single-cell datasets using self-supervised learning objectives, enabling them to be adapted to a wide range of downstream biological tasks [1]. Inspired by the remarkable success of large language models (LLMs) in natural language processing, researchers have developed scFMs that treat individual cells as "sentences" and genes or genomic features as "words" or "tokens" in a cellular language [1] [2]. This approach has fundamentally transformed our ability to interpret the complex language of gene regulation, cellular states, and biological systems at single-cell resolution.
The development of scFMs addresses a critical need in single-cell genomics for unified frameworks capable of integrating and comprehensively analyzing rapidly expanding data repositories [1]. With public archives now containing tens of millions of single-cell omics datasets spanning diverse cell types, states, and conditions, traditional analytical approaches struggle to leverage this wealth of information effectively [1]. scFMs overcome these limitations by learning fundamental principles of cellular biology from millions of cells encompassing many tissues and conditions, capturing biological variation at an unprecedented scale. This knowledge can then be transferred to new datasets or downstream tasks through fine-tuning or zero-shot learning approaches [3], establishing scFMs as pivotal tools for advancing our understanding of cellular function and disease mechanisms.
Fundamental Concepts: From Natural Language to Cellular Linguistics
Architectural Foundations: Transformers in Biology
The transformer architecture, which has revolutionized natural language processing and computer vision, serves as the computational backbone of single-cell foundation models [1] [4]. Transformers are neural network architectures characterized by attention mechanisms that allow the model to learn and weight relationships between any pair of input tokens [1]. In the context of single-cell biology, this attention mechanism enables scFMs to determine which genes in a cell are most informative of the cell's identity or state, how they co-vary across cells, and how they participate in regulatory or functional networks [1].
Most scFMs employ variants of the transformer architecture, primarily falling into two categories: encoder-based models and decoder-based models [1]. Encoder-based models, such as those inspired by BERT (Bidirectional Encoder Representations from Transformers), utilize bidirectional attention mechanisms where the model learns from the context of all genes in a cell simultaneously [1] [5]. In contrast, decoder-based models like scGPT use a unidirectional masked self-attention mechanism that iteratively predicts masked genes conditioned on known genes [1] [4]. Each architectural approach offers distinct advantages—encoder models typically excel at classification and embedding tasks, while decoder models show stronger performance in generation tasks—though no single architecture has emerged as clearly superior for single-cell data [1].
The Cellular Linguistics Framework
The core conceptual innovation underlying scFMs is the treatment of cellular biology as a language with its own grammar and semantics. In this framework, individual cells are treated analogously to sentences, while genes or genomic features along with their expression values serve as words or tokens [1] [2]. This analogy enables the application of linguistic principles to biological data, where cellular states can be "read" and "interpreted" through their gene expression patterns, much like sentences can be understood through their constituent words.
The premise of this approach is that by exposing a model to millions of cells encompassing many tissues and conditions, the model can learn the fundamental "grammar" of cellular behavior—the rules governing how genes interact and coordinate their expression to define cell identity and function [1]. This learned knowledge enables scFMs to generalize to new biological contexts and perform various analytical tasks without task-specific training, mirroring the zero-shot capabilities of large language models [3]. The cellular linguistics framework thus provides a powerful conceptual bridge between natural language understanding and biological interpretation, enabling researchers to decipher the complex language of cellular systems.
Technical Architecture of Single-Cell Foundation Models
Data Tokenization and Encoding Strategies
Tokenization, the process of converting raw input data into discrete units called tokens, represents a critical challenge in adapting transformer architectures to single-cell data. Unlike natural language, where words have inherent sequential order, gene expression data lacks natural ordering, requiring specialized tokenization strategies [1] [3].
Table: Tokenization and Encoding Strategies in Single-Cell Foundation Models
| Component | Encoding Method | Description | Example Models |
|---|---|---|---|
| Gene Identity | Learnable embedding | Each gene projected into high-dimensional space via one-hot encoding + projection network | scBERT, scGPT, Geneformer |
| External knowledge integration | Incorporates promoter embeddings, co-expression patterns, or protein language model outputs | GeneCompass, UCE | |
| Expression Values | Rank encoding | Genes sorted by expression level; position encoded via positional encoding | Geneformer, tGPT |
| Continuous value encoding | Direct projection of continuous expression values into embedding space | scFoundation, GeneCompass | |
| Discrete value encoding | Expression values discretized into bins; each bin treated as categorical variable | scGPT, scMulan | |
| Reference encoding | Expression values used as sampling weights or reference for gene embeddings | UCE, scELMo | |
| Extra Information | Modality tokens | Special tokens indicating data type (e.g., scRNA-seq, scATAC-seq) | scGPT, Nicheformer |
| Batch tokens | Tokens representing batch information to address technical variability | scGPT | |
| Metadata incorporation | Integration of cell metadata, spatial coordinates, or experimental conditions | scMulan, Nicheformer |
A fundamental challenge in applying transformers to single-cell data is that gene expression data are not naturally sequential [1] [3]. Unlike words in a sentence, genes in a cell have no inherent ordering, requiring researchers to impose artificial structure. Common strategies include ranking genes within each cell by their expression levels and feeding the ordered list of top genes as the "sentence" [1]. Other models partition genes into bins by their expression values or simply use normalized counts without complex ranking schemes [1]. Each gene is typically represented as a composite token embedding that combines a gene identifier and its expression value in the given cell, with positional encoding schemes adapted to represent the relative order or rank of each gene [1].
Model Architectures and Pretraining Strategies
scFMs employ diverse architectural configurations and pretraining objectives tailored to single-cell data. The transformer backbone processes tokenized gene expression data through multiple layers of self-attention and feed-forward networks, gradually building latent representations of cells and genes [1].
Table: Architecture and Pretraining Approaches in Representative Single-Cell Foundation Models
| Model | Architecture Type | Pretraining Data Scale | Pretraining Objective | Special Features |
|---|---|---|---|---|
| Geneformer | BERT-like encoder | 30 million cells | Masked gene prediction | Rank-based encoding; network biology focus |
| scGPT | GPT-like decoder | 33 million human cells | Masked gene prediction | Multi-omic support; discrete value encoding |
| scBERT | Performer encoder | Not specified | Cell type prediction | Focus on cell type annotation |
| scFoundation | Transformer encoder | 50 million cells | Masked gene prediction | Continuous value encoding |
| Nicheformer | Hybrid transformer | 110 million cells | Multi-task learning | Integrates single-cell + spatial data |
| scPlantLLM | Transformer | Plant-specific data | Masked LM + cell annotation | Specialized for plant genomics |
Pretraining scFMs involves self-supervised learning on massive single-cell datasets without explicit labeling [1]. The most common pretraining objective is masked language modeling, where random subsets of genes are masked from the input, and the model is trained to predict the masked genes based on the remaining context [1] [5]. This approach forces the model to learn the complex dependencies and relationships between genes, effectively capturing the underlying "grammar" of gene regulation. Through this process, scFMs develop rich internal representations that encode biological knowledge about cellular states, gene functions, and regulatory relationships, which can then be transferred to various downstream tasks through fine-tuning or used directly in zero-shot settings [3].
Experimental Framework and Benchmarking
Benchmarking Methodologies for scFM Evaluation
Comprehensive benchmarking is essential for evaluating the performance and capabilities of scFMs. Recent studies have developed sophisticated evaluation frameworks that assess models across multiple dimensions, including biological relevance, technical performance, and practical utility [3]. These benchmarks typically evaluate scFMs against well-established baseline methods under realistic conditions across diverse tasks, such as batch integration, cell type annotation, cancer cell identification, and drug sensitivity prediction [3].
Evaluation metrics for scFMs span unsupervised, supervised, and knowledge-based approaches [3]. Traditional metrics assess technical performance like clustering accuracy and batch correction effectiveness, while novel biologically-grounded metrics evaluate the ability of models to capture meaningful biological relationships. These include scGraph-OntoRWR, which measures the consistency of cell type relationships captured by scFMs with prior biological knowledge, and Lowest Common Ancestor Distance (LCAD), which assesses the severity of errors in cell type annotation based on ontological proximity [3]. Such biologically-informed metrics provide crucial insights into whether scFMs capture functionally relevant biological patterns beyond technical optimizations.
Key Experimental Protocols
Zero-Shot Evaluation Protocol
The zero-shot capabilities of scFMs represent one of their most powerful features, enabling model assessment without task-specific training [3]. The standard zero-shot evaluation protocol involves:
- Embedding Extraction: Generating cell or gene embeddings from pretrained models without any fine-tuning
- Task Application: Applying these embeddings to specific downstream tasks (e.g., clustering, classification)
- Performance Assessment: Evaluating performance using task-appropriate metrics compared to baseline methods
This protocol tests the general biological knowledge encoded during pretraining and the model's ability to transfer this knowledge to novel tasks and datasets [3]. Studies have shown that scFMs pretrained on diverse cellular contexts can generate embeddings that capture meaningful biological relationships, enabling effective performance even on previously unseen cell types or conditions [3] [6].
Cross-Species and Cross-Tissue Generalization Assessment
A critical test for scFMs is their ability to generalize across biological contexts not seen during training. The standard protocol involves:
- Model Pretraining: Training models on data from specific species or tissues
- Cross-Context Evaluation: Applying models to data from different species or tissue types
- Performance Comparison: Assessing whether performance degrades gracefully or maintains effectiveness
This evaluation has revealed that while some models show remarkable cross-context generalization capabilities (e.g., scPlantLLM performing zero-shot learning on unseen plant species) [6], performance varies significantly across models and tasks, highlighting the importance of dataset diversity during pretraining.
The development and application of scFMs rely on curated data resources that provide standardized, high-quality single-cell datasets.
Table: Key Data Resources for Single-Cell Foundation Models
| Resource Name | Scale | Content Description | Primary Use in scFMs |
|---|---|---|---|
| CZ CELLxGENE | 100+ million cells | Annotated single-cell datasets from diverse tissues and conditions | Primary pretraining corpus for many models |
| Human Cell Atlas | Not specified | Comprehensive reference map of all human cells | Pretraining data source |
| PanglaoDB | Not specified | Curated compendium of single-cell data | Pretraining and benchmarking |
| SpatialCorpus-110M | 110 million cells | Curated single-cell + spatial transcriptomics data | Pretraining for spatial-aware models |
| DISCO | Not specified | Single-cell data across human tissues and development | Multitask pretraining |
| Asian Immune Diversity Atlas (AIDA) | Not specified | Diverse human immune cell data | Benchmarking and validation |
Computational Infrastructure and Software Tools
Implementing scFMs requires substantial computational resources and specialized software tools. The transformer architectures used in these models typically require graphical processing units (GPUs) with substantial memory (16GB+) for both training and inference [1] [2]. Training large-scale scFMs from scratch may require hundreds or thousands of GPU hours distributed across multiple high-end processors [1], though fine-tuning pretrained models for specific tasks is computationally less intensive.
Key software frameworks for developing and applying scFMs include PyTorch and TensorFlow for model implementation, Hugging Face Transformers for architectural components, and specialized single-cell analysis libraries like Scanpy for data preprocessing and evaluation [7]. The field is also developing standardized benchmarking platforms such as scBench [3] to facilitate fair comparison across different models and methods.
Signaling Pathways and Biological Workflows
scFM Analytical Workflow
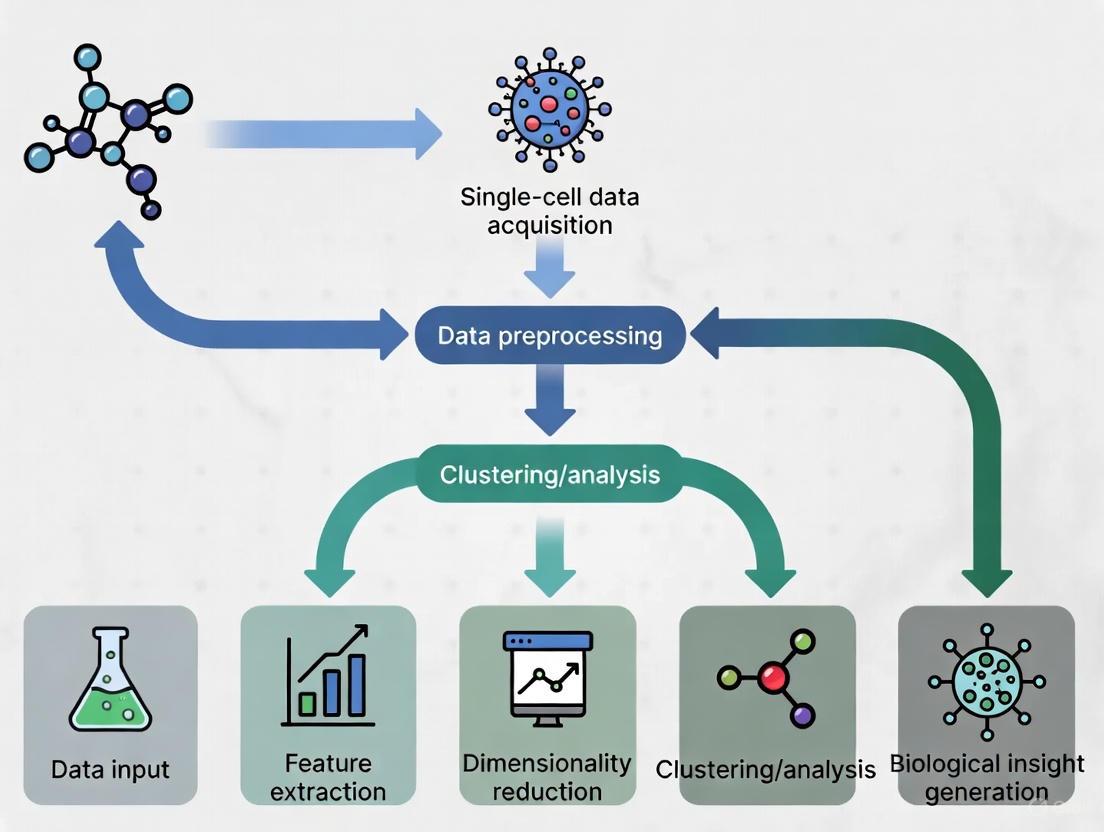
The following diagram illustrates the standard analytical workflow for applying single-cell foundation models to biological research questions:
Multi-modal scFM Architecture
The following diagram illustrates the architecture of a multi-modal single-cell foundation model capable of integrating diverse data types:
Performance Benchmarking and Comparative Analysis
Quantitative Performance Across Tasks
Rigorous benchmarking studies have evaluated scFMs across diverse biological and clinical tasks to assess their practical utility and performance advantages over traditional methods.
Table: Performance Comparison of Single-Cell Foundation Models Across Key Tasks
| Task Category | Best Performing Model(s) | Key Performance Metrics | Advantage Over Baseline |
|---|---|---|---|
| Cell Type Annotation | scBERT, scGPT | Accuracy: 85-95% (varies by dataset) | Superior for novel/rare cell types |
| Batch Integration | scGPT, scVI | LISI scores: 1.5-2.5 (higher better) | Better biological preservation |
| Drug Sensitivity Prediction | Geneformer, scGPT | AUROC: 0.75-0.90 | Context-aware predictions |
| Spatial Pattern Reconstruction | Nicheformer | Spatial correlation: 0.6-0.8 | Transfer spatial context to dissociated cells |
| Cross-Species Generalization | scPlantLLM, UCE | Zero-shot accuracy: 70-85% | Leverage protein language models |
| Perturbation Prediction | Geneformer, scGPT | Top-k accuracy: 0.7-0.9 | Identify master regulators |
Benchmarking results reveal that no single scFM consistently outperforms all others across every task, emphasizing the importance of task-specific model selection [3]. While scFMs generally demonstrate robust performance across diverse applications, simpler machine learning models can sometimes outperform foundation models on specific tasks, particularly under resource constraints or with limited data [3]. This highlights that the choice between complex foundation models and simpler alternatives should be guided by factors including dataset size, task complexity, need for biological interpretability, and available computational resources [3].
Biological Insight Extraction
Beyond technical performance metrics, scFMs are evaluated on their ability to generate novel biological insights. Models are assessed through attention-based interpretability analyses that examine which genes the model attends to when making specific predictions [3]. For example, studies have shown that scFMs can identify biologically meaningful gene-gene relationships and regulatory networks without explicit supervision, demonstrating that these models capture functionally relevant biological patterns [3].
The biological relevance of scFM embeddings is further validated through gene-level tasks that assess whether functionally similar genes are embedded in close proximity in the latent space [3]. Performance on predicting known biological relationships, including tissue specificity and Gene Ontology terms, provides crucial evidence that scFMs learn biologically meaningful representations rather than merely technical artifacts of the data [3].
Future Directions and Challenges
Despite their significant promise, scFMs face several important challenges that represent active areas of research. A primary limitation is the non-sequential nature of omics data, which complicates the direct application of transformer architectures designed for sequential data [1]. Additional challenges include inconsistency in data quality across studies, the computational intensity required for training and fine-tuning, and difficulty in interpreting the biological relevance of latent embeddings and model representations [1].
Future development of scFMs will likely focus on several key directions. Multi-modal integration represents a frontier, with models increasingly incorporating diverse data types including transcriptomics, epigenomics, proteomics, and cellular images [6] [8]. Spatial context awareness is another critical direction, with models like Nicheformer pioneering the integration of spatial relationships into cellular representations [8]. Additionally, efforts to improve model interpretability and biological grounding will be essential for building trust and facilitating biological discovery [3]. Finally, development of more efficient architectures and training methods will be crucial for making scFMs accessible to researchers with limited computational resources [1] [3].
As the field progresses, scFMs are poised to become increasingly central to single-cell research, potentially evolving toward comprehensive "virtual cell" models that can simulate cellular behavior and response in silico [8]. This trajectory promises to deepen our understanding of cellular biology and accelerate therapeutic development across a wide range of diseases.
Single-cell foundation models (scFMs) represent a paradigm shift in computational biology, leveraging the power of transformer networks to decipher the complex language of cellular function. Inspired by their success in natural language processing (NLP), these models are pretrained on vast datasets comprising millions of single cells to learn universal biological representations [1] [9]. The core premise is that individual cells can be treated as sentences, with genes and their expression levels acting as the words or tokens [1] [2]. This adaptation allows transformers to capture intricate patterns of gene-gene interactions and cellular heterogeneity, providing a foundational tool for a wide range of downstream biological tasks, from cell type annotation to perturbation prediction [1] [9] [3].
Core Architectural Framework
Transformer Architecture: From NLP to Single-Cell Biology
The transformer architecture, characterized by its self-attention mechanism, forms the backbone of modern scFMs. Self-attention allows the model to dynamically weigh and consider the relationships between all genes in a cell simultaneously, thereby capturing complex, non-linear dependencies within the gene expression profile [1]. In biological terms, this enables the model to learn how the expression of one gene might influence or be associated with the expression of thousands of others across diverse cellular contexts [1] [3].
Most scFMs implement specific variants of the transformer architecture:
- Encoder-based models (e.g., BERT-like): These models use a bidirectional attention mechanism, meaning each gene token can attend to all other genes in the cell context. This is particularly useful for tasks that require a comprehensive understanding of the entire cellular state, such as cell type classification [1].
- Decoder-based models (e.g., GPT-like): These models employ a unidirectional or masked self-attention mechanism, where a gene can only attend to preceding genes in the sequence. This architecture is often used for generative tasks, such as predicting masked genes or in-silico perturbation effects [1].
- Hybrid designs: Some models explore custom combinations of encoder and decoder blocks, though a clearly superior architecture for single-cell data has not yet emerged [1].
Key Adaptation: Tokenization Strategies for Single-Cell Data
A critical challenge in adapting transformers to single-cell omics is tokenization—the process of converting raw gene expression data into a sequence of discrete tokens that the model can process. Unlike words in a sentence, genes lack a natural sequential order [1] [3]. To overcome this, several strategies have been developed, as summarized in the table below.
Table 1: Common Tokenization Strategies in Single-Cell Foundation Models
| Strategy | Description | Rationale | Examples of Use |
|---|---|---|---|
| Rank-based Encoding | Genes are ordered by their expression level within each cell, from highest to lowest. | Provides a deterministic, cell-specific sequence that reflects the most to least active genes. | scGPT [1], Nicheformer [10], Geneformer [10] |
| Binning | Expression values are partitioned into discrete bins or value ranges, and each bin is treated as a token. | Reduces the complexity of continuous expression values and can capture nonlinear relationships. | scBERT [1] |
| Normalized Counts | Uses normalized expression counts (e.g., log-transformed) directly or with minimal discretization. | Simplicity; avoids potential information loss from aggressive binning or ranking. | Some models report no advantage to complex ranking [1] |
The tokenization process typically results in a sequence where each gene is represented by an embedding vector that combines a gene identifier embedding (analogous to a word embedding) and a value embedding (representing its expression level) [3]. To provide the model with structural information, positional encodings are added to inform the model of the gene's rank or position in the input sequence [1] [2].
Furthermore, special tokens are often incorporated to enrich the biological context:
- Cell-level context tokens: Prepend information about the cell's identity or metadata [1].
- Modality tokens: Indicate the source of the data (e.g., scRNA-seq, scATAC-seq, spatial transcriptomics) [1] [10].
- Batch tokens: Used to account for technical variations between different experiments [1].
The following diagram illustrates the complete tokenization and data preparation workflow for a transformer model like scGPT or Nicheformer.
Comparative Analysis of Model Architectures
The field has seen the development of several prominent scFMs, each with distinct architectural choices and pretraining corpora. The table below provides a quantitative comparison of key models.
Table 2: Comparative Analysis of Single-Cell Foundation Models
| Model Name | Core Architecture | Pretraining Data Scale | Key Technical Features | Notable Applications |
|---|---|---|---|---|
| scGPT [1] [9] | Decoder (GPT-like) | 33+ million cells [9] | Uses rank-based gene tokenization; focuses on generative tasks. | Zero-shot cell annotation, multi-omic integration, in-silico perturbation prediction. |
| Geneformer [10] [3] | Encoder (BERT-like) | Not specified in detail | Employs rank-based encoding; context length of 2,042 genes. | Cell network inference, disease module identification. |
| Nicheformer [10] | Encoder | 110 million cells (SpatialCorpus-110M) | Jointly trained on dissociated and spatial transcriptomics data; uses species and technology tokens. | Spatial composition prediction, transferring spatial context to dissociated data. |
| scBERT [1] | Encoder (BERT-like) | Millions of cells | Uses gene binning for tokenization. | Cell type annotation. |
| UCE [3] | Encoder | Not specified in detail | A unified cell embedding model. | Cell type annotation, dataset integration. |
Experimental Protocols and Methodologies
Pretraining Strategies
The power of scFMs stems from self-supervised pretraining on massive, diverse collections of single-cell data. The primary objective is to learn generalizable representations of gene and cell function without the need for human-annotated labels [1] [10].
A. Data Sourcing and Curation:
- Sources: Pretraining corpora are assembled from public repositories such as the Gene Expression Omnibus (GEO), Sequence Read Archive (SRA), CZ CELLxGENE, the Human Cell Atlas, and PanglaoDB [1].
- Curation: This involves careful selection of datasets, filtering of low-quality cells and genes, and balancing dataset compositions to capture a wide spectrum of biological variation across tissues, species, and disease states [1]. A key challenge is managing batch effects and technical noise from different experimental protocols [1] [10].
B. Self-Supervised Pretraining Tasks:
- Masked Gene Modeling: A fraction (e.g., 15-20%) of the input gene tokens are randomly masked or replaced. The model is then trained to predict the original identity and/or expression value of the masked genes based on the context provided by the unmasked genes [1]. This task forces the model to learn the complex co-expression relationships and dependencies between genes.
- Contrastive Learning: Used in some models to align representations of similar cells or genes while pushing apart representations of dissimilar ones, often leveraging data augmentations [9].
Model Fine-Tuning and Evaluation
After pretraining, scFMs can be adapted to specific downstream tasks through fine-tuning or linear probing.
A. Fine-Tuning: The entire model or a subset of its layers is further trained on a smaller, task-specific labeled dataset. This process adjusts the model's parameters to specialize for the target application [1] [10].
B. Linear Probing: The weights of the pretrained model are frozen, and only a simple linear classifier is trained on top of the extracted cell or gene embeddings. This method tests the quality and generalizability of the representations learned during pretraining [10] [3].
C. Key Downstream Tasks for Evaluation:
- Cell-level tasks: Cell type annotation, batch integration, and patient classification [3].
- Gene-level tasks: Gene function prediction, gene regulatory network (GRN) inference, and analysis of gene-gene relationships [3].
- Spatial tasks: Prediction of spatial cellular niches and tissue region composition (for spatially-aware models like Nicheformer) [10].
- Perturbation modeling: Predicting cellular responses to genetic or chemical perturbations in silico [1] [2].
The Scientist's Toolkit
The following table details key computational reagents and resources essential for working with single-cell foundation models.
Table 3: Essential Research Reagents and Resources for scFM Research
| Item / Resource | Function / Description | Example Tools / Platforms |
|---|---|---|
| Pretrained Model Weights | The learned parameters of a scFM, allowing researchers to perform inference or fine-tuning without the prohibitive cost of pretraining. | scGPT, Geneformer, Nicheformer model checkpoints [1] [10]. |
| Processed Data Corpora | Large-scale, curated collections of single-cell data used for model pretraining and benchmarking. | CZ CELLxGENE [1] [9], DISCO [9], SpatialCorpus-110M [10]. |
| Benchmarking Frameworks | Standardized platforms for evaluating and comparing the performance of different scFMs across a suite of biological tasks. | BioLLM [9], custom benchmarking pipelines [3]. |
| Visualization Tools | Software for exploring and interpreting single-cell data and model outputs, such as embeddings and attention weights. | scViewer [11], cellxgene [11], UCSC Cell Browser [11]. |
Visualizing the End-to-End Workflow
The entire process, from data preparation to model output and application, is summarized in the following workflow diagram.
Single-cell foundation models (scFMs) are revolutionizing our understanding of cellular biology and disease by leveraging large-scale, self-supervised learning. The performance and capability of these models are intrinsically linked to the scale and quality of their training data. This technical guide explores the central role of massive public repositories, with a focus on CZ CELLxGENE, in constructing the foundational corpora that power scFMs, enabling them to decode the complex "language" of gene regulation and cellular function for biomedical research and drug discovery.
The Indispensable Role of Large-Scale Data in scFM Development
A foundation model is defined as a large-scale deep learning model pretrained on vast datasets, which can then be adapted to a wide range of downstream tasks. The success of this paradigm in single-cell biology is contingent on the availability of extensive and diverse training corpora [1].
The public domain now contains tens of millions of single-cell omics datasets, spanning a vast array of cell types, states, and conditions. This wealth of data enables researchers to train large models to decipher the fundamental principles of cellular behavior. The premise is that by exposing a model to millions of cells from diverse tissues and conditions, it can learn generalizable representations that transfer effectively to new datasets or tasks, such as cell type annotation, perturbation prediction, and disease state classification [1]. The performance of these models has been shown to scale predictably with both the volume of pre-training data and the number of model parameters, making repositories like CELLxGENE critical for state-of-the-art performance [12].
Public databases aggregate and curate single-cell data, providing the essential raw material for scFM training. The table below summarizes key repositories, highlighting their scale and specialization.
Table 1: Key Public Single-Cell RNA-Sequencing Databases and Their Scope
| Database Name | Description | Scale (Number of Cells) | Primary Focus |
|---|---|---|---|
| CZ CELLxGENE [13] | A platform for downloading and visually exploring single-cell data. | >33 million | General, multi-species |
| Arc Virtual Cell Atlas [14] | An AI-curated repository integrating single-cell profiles. | >300 million | General, multi-species |
| DISCO [14] | A database aggregating and harmonizing public single-cell datasets. | >100 million | General |
| Human Cell Atlas (HCA) [14] | A global collaborative effort to create comprehensive reference maps of all human cells. | 58 million | General (Human) |
| PanglaoDB [14] | A database of mouse and human scRNA-seq experiments with pre-annotated cell-type markers. | Information Missing | General |
| Tumor Immune Single-cell Hub (TISCH2) [14] | A resource dedicated to the tumor microenvironment. | Information Missing | Cancer-focused |
These resources provide the immense sample sizes needed to power scFMs. For example, the Teddy family of foundation models was trained on a corpus of 116 million cells sourced directly from CELLxGENE, leading to substantial improvements in downstream tasks like disease state identification [12]. The integration of many studies within these databases provides enormous aggregate cell counts, boosting statistical power for detecting rare cell populations or subtle gene expression changes that would be impossible to discern in individual studies [14].
CELLxGENE: A Cornerstone for scFM Pretraining
CELLxGENE has emerged as a critical infrastructure component for the single-cell research community. It provides unified access to a massive, standardized corpus of single-cell data, which is a prerequisite for effective model training.
The platform provides over 33 million unique cells from more than 436 datasets, encompassing over 2,700 cell types from human and mouse tissues [13]. This diversity is crucial for training models that can generalize across biological contexts. CELLxGENE facilitates model development not just as a data source but also as a platform for community innovation, showcasing research projects that leverage its data, such as PINNACLE (a model for contextual protein biology) and scCIPHER (a deep learning approach for precision medicine in neurological disorders) [15].
Table 2: Computational Tools and Resources for scFM Research
| Tool / Resource | Type | Primary Function in scFM Workflow |
|---|---|---|
| CELLxGENE Census [13] | Python/R API | Provides programmatic access to any custom slice of standardized cell data from the CELLxGENE corpus for model training and analysis. |
| Seurat [16] | Software Toolkit | A comprehensive toolkit for single-cell analysis, often used as a baseline or for comparison in reference mapping and data integration tasks. |
| Harmony [3] | Algorithm | A clustering-based method for dataset integration, used to correct for batch effects while preserving biological variation. |
| scVI [3] | Generative Model | A probabilistic neural network for single-cell data integration, used as a baseline model in benchmarking studies against scFMs. |
| Scanpy [14] | Python Library | A scalable toolkit for analyzing single-cell gene expression data, commonly used for preprocessing data before model training. |
A Technical Workflow for Leveraging CELLxGENE in scFM Development
The process of transforming raw data from a repository like CELLxGENE into a trained scFM involves several critical, interconnected steps. The following diagram illustrates this end-to-end workflow.
Data Curation and Assembly
The first step involves the careful selection and integration of datasets from the CELLxGENE corpus to create a pretraining cohort. This step is as important as the model architecture itself [1]. Challenges include managing batch effects, technical noise from different sequencing protocols, and varying data processing steps [1]. Effective pretraining requires meticulous dataset selection, filtering of cells and genes, balancing dataset compositions, and rigorous quality control [1]. CELLxGENE aids this process by providing standardized data and metadata, which reduces the preprocessing burden on model developers.
Tokenization and Input Representation
Tokenization converts raw gene expression data into a sequence of discrete units (tokens) that the model can process. This is a critical and non-trivial step because, unlike words in a sentence, genes have no inherent sequential order [1]. Common strategies implemented by scFMs include:
- Rank-based Encoding: Genes within each cell are ranked by their expression levels, and the ordered list of top genes is treated as the "sentence" representing the cell. This is used by models like Geneformer and Teddy-G [1] [12].
- Binning: Gene expression values are discretized into bins, and the model is trained to predict the bin for a given gene. This approach is used by scGPT and the Teddy-X variant [1] [12].
- Value Embeddings: Some models, like scFoundation, represent gene expressions as weighted combinations of learned embeddings [12].
During this step, gene metadata (e.g., gene ontology terms) and cell metadata (e.g., tissue of origin) can be incorporated as special tokens to provide richer biological context for the model [1].
Model Pretraining and Fine-Tuning
After tokenization, models are pretrained on a self-supervised task using the entire curated corpus. The most common objective is Masked Language Modeling (MLM), where a random subset of gene tokens is masked, and the model is trained to predict them based on the context of the unmasked genes in the cell [1] [12]. This process forces the model to learn the underlying relationships and co-dependencies between genes.
Once a model is pretrained, it possesses a general understanding of cellular biology. This base model can then be efficiently fine-tuned with a smaller, task-specific dataset for downstream applications such as cell type annotation, drug sensitivity prediction, or disease classification [3]. This "pre-train then fine-tune" paradigm allows the knowledge gained from millions of cells to be transferred to specialized tasks with limited labeled data.
Experimental Validation and Benchmarking
To validate the effectiveness of scFMs trained on large repositories, researchers conduct rigorous benchmarking studies. A 2025 benchmark evaluated six scFMs against established baselines on both biological and clinically relevant tasks [3]. The findings reveal that scFMs are robust and versatile but also highlight important nuances:
- Robustness and Versatility: scFMs demonstrate strong performance across diverse tasks, including dataset integration and cell type annotation, particularly in preserving biological relationships as measured by novel ontology-informed metrics [3].
- No Single Best Model: No single scFM consistently outperformed all others across every task. Model selection must be tailored based on factors like dataset size, task complexity, and computational resources [3].
- Comparison to Simpler Models: In certain scenarios, simpler, task-specific machine learning models can be more efficient and perform on par with or even surpass large foundation models, especially when computational resources are constrained [3] [12].
These results underscore that while large-scale data is powerful, its effective translation into model performance depends on thoughtful architectural choices and training strategies.
The rapid accumulation of single-cell genomics data has created an unprecedented opportunity to decipher the fundamental principles of cellular function. Drawing inspiration from the success of large language models (LLMs) in natural language processing (NLP), researchers have begun treating cellular biology as a linguistic system, creating single-cell foundation models (scFMs) that reinterpret biological data through a computational linguistic lens [1]. In this analogous framework, individual cells are treated as "sentences" while genes and their expression values become the "words" or tokens that constitute these sentences [1] [6]. This paradigm shift enables the application of transformer-based architectures, which have revolutionized NLP, to the complex, high-dimensional space of single-cell data, potentially unlocking deeper insights into cellular heterogeneity, gene regulatory networks, and disease mechanisms [1].
The core premise of this approach is that by exposing a model to millions of cells encompassing diverse tissues, species, and biological conditions, the model can learn the fundamental "grammar" and "syntax" of cellular behavior [1]. Just as LLMs learn contextual relationships between words by training on vast text corpora, scFMs learn the contextual relationships between genes across different cellular states and environments [3]. This whitepaper explores the technical foundations, methodological considerations, and practical applications of this transformative analogy, providing researchers with a comprehensive guide to understanding and utilizing single-cell foundation models in biological and pharmaceutical research.
Core Architectural Framework
Tokenization Strategies for Single-Cell Data
Tokenization converts raw gene expression data into discrete units (tokens) that models can process. Unlike natural language, where words have inherent sequence, gene expression data lacks natural ordering, presenting a fundamental challenge for applying sequential models [1]. Researchers have developed several strategic approaches to address this challenge, as summarized in Table 1.
Table 1: Tokenization Strategies in Single-Cell Foundation Models
| Strategy | Method | Advantages | Limitations |
|---|---|---|---|
| Expression Ranking | Genes are ranked by expression levels within each cell; top genes form the "sentence" [1] | Deterministic; provides structured input for transformers | Arbitrary sequence may not reflect biological gene relationships |
| Value Binning | Expression values are partitioned into bins; bin assignments determine token identity [1] | Captures expression magnitude information; reduces vocabulary size | May lose fine-grained expression differences |
| Normalized Counts | Uses normalized expression values directly without complex ranking [1] | Simpler implementation; maintains continuous nature of data | Requires careful normalization to handle technical variability |
| Multi-Modal Tokens | Incorporates special tokens for different omics modalities (e.g., ATAC-seq, proteomics) [1] | Enables integrated multi-omics analysis; captures broader regulatory context | Increases model complexity and computational requirements |
In addition to these core tokenization methods, models often incorporate special tokens to enrich biological context. These may include tokens representing cell identity metadata, batch information, or gene metadata such as chromosomal location or Gene Ontology terms [1]. After tokenization, all tokens are converted to embedding vectors processed by transformer layers, ultimately producing latent embeddings for each gene token and often a dedicated embedding for the entire cell [1].
Model Architectures and Attention Mechanisms
Most scFMs utilize transformer architectures characterized by attention mechanisms that learn and weight relationships between any pair of input tokens [1]. These attention mechanisms enable models to determine which genes in a cell are most informative of the cell's identity or state, how genes covary across cells, and how they participate in regulatory or functional connections [1]. The gene expression profile of each cell is converted to a set of gene tokens that serve as inputs, and the attention layers gradually build latent representations of each cell and gene [1].
Two predominant architectural paradigms have emerged in scFM development, each with distinct characteristics and applications, as detailed in Table 2.
Table 2: Transformer Architectures in Single-Cell Foundation Models
| Architecture | Attention Mechanism | Common Applications | Representative Models |
|---|---|---|---|
| BERT-like Encoder | Bidirectional attention; learns from all genes in a cell simultaneously [1] | Cell type annotation; embedding generation; classification tasks | scBERT [1] |
| GPT-like Decoder | Unidirectional masked self-attention; iteratively predicts masked genes conditioned on known genes [1] | Generative tasks; perturbation prediction; sequence generation | scGPT [1] |
| Encoder-Decoder | Combines both bidirectional and unidirectional attention; can encode input and decode output [1] | Multi-task learning; complex prediction tasks | Hybrid designs under exploration [1] |
The attention mechanism in scFMs can be visualized as a process where each gene "looks" at other genes in the same cell to determine their contextual relationships. This process generates attention weights that signify the strength of relationships between gene pairs, potentially mirroring biological interactions such as co-regulation or functional pathway membership [3].
Figure 1: Core Architecture of Single-Cell Foundation Models
Experimental Protocols and Benchmarking
Pretraining Strategies and Self-Supervised Learning
Pretraining scFMs involves training on self-supervised tasks across unlabeled single-cell data, typically using masked language modeling objectives [1]. In this approach, random subsets of gene tokens are masked, and the model is trained to predict the masked tokens based on the context provided by the remaining genes in the cell [1]. This process enables the model to learn the fundamental relationships between genes and cellular states without requiring manually annotated labels.
The scale of pretraining corpora has expanded dramatically, with recent models training on datasets containing tens to hundreds of millions of cells from diverse sources including CELLxGENE, Human Cell Atlas, and GEO [1] [17]. For example, the CellWhisperer framework was trained on over 1 million human RNA-seq profiles with matched textual annotations created through LLM-assisted curation of sample metadata [17]. This massive scale is essential for capturing the broad spectrum of biological variation across cell types, tissues, and conditions.
Benchmarking Framework and Performance Metrics
Comprehensive benchmarking studies have evaluated scFMs against traditional methods to assess their capabilities and limitations. A recent biology-driven benchmark evaluated six scFMs against established baselines across two gene-level and four cell-level tasks using 12 different metrics [3]. The evaluation encompassed both pre-clinical applications (batch integration and cell type annotation across five datasets with diverse biological conditions) and clinically relevant tasks (cancer cell identification and drug sensitivity prediction across seven cancer types and four drugs) [3].
Table 3 summarizes key benchmarking results that highlight the comparative performance of scFMs versus traditional methods across different task categories.
Table 3: Performance Comparison of scFMs vs. Traditional Methods
| Task Category | Superior Approach | Key Findings | Practical Implications |
|---|---|---|---|
| Batch Integration | scFMs show advantages in large, complex datasets [18] | Deep learning methods better preserve biological variation while removing technical artifacts | Recommended for atlas-level integration projects |
| Cell Type Annotation | scFMs excel in zero-shot learning for novel cell types [3] | Foundation models transfer knowledge to unseen cell types better than traditional classifiers | Valuable for discovering or characterizing rare cell populations |
| Drug Sensitivity Prediction | Traditional ML can outperform for specific, narrow datasets [3] | Simpler models may adapt more efficiently to homogeneous data with limited samples | Consider task specificity and data resources when selecting approach |
| Gene Function Prediction | scFMs provide biologically meaningful gene embeddings [3] | Gene embeddings capture functional relationships and tissue specificity | Useful for inferring gene function and regulatory relationships |
Notably, benchmarking reveals that no single scFM consistently outperforms others across all tasks, emphasizing the need for tailored model selection based on factors such as dataset size, task complexity, biological interpretability requirements, and computational resources [3]. To address this challenge, researchers have proposed the roughness index (ROGI) as a proxy metric to recommend appropriate models in a dataset-dependent manner [3].
Visualization of Model Training and Evaluation Workflow
Figure 2: scFM Training and Evaluation Workflow
Implementing and utilizing single-cell foundation models requires familiarity with both computational resources and biological data sources. Table 4 provides a comprehensive overview of key tools, platforms, and datasets essential for researchers working with scFMs.
Table 4: Essential Research Reagents and Resources for scFM Research
| Resource Category | Specific Examples | Function and Utility | Access Information |
|---|---|---|---|
| Data Repositories | CELLxGENE [1], GEO [17], Human Cell Atlas [1] | Provide standardized, annotated single-cell datasets for model training and validation | Publicly available; CELLxGENE contains >100 million unique cells [1] |
| Pre-trained Models | scGPT [1], Geneformer [3], scPlantLLM [6] | Offer transfer learning capabilities; can be fine-tuned for specific applications without pretraining from scratch | Various licensing; some open-source implementations available |
| Benchmarking Frameworks | scIB [18], Biology-Driven Benchmark [3] | Provide standardized evaluation metrics and pipelines for comparing model performance | Open-source implementations; custom metrics for biological relevance |
| Specialized Tools | CellWhisperer [17], scANVI [18] | Enable specific applications like natural language query or semi-supervised integration | Mixed availability; some open-source, some proprietary |
| Computational Infrastructure | GPU clusters, Cloud computing platforms | Handle intensive computational requirements of training and fine-tuning large foundation models | Institutional HPC, commercial cloud providers |
Specialized domain-adapted models have also emerged to address specific research contexts. For example, scPlantLLM represents a transformer-based model specifically trained on plant single-cell data to address unique challenges such as polyploidy, cell walls, and complex tissue-specific expression patterns not adequately handled by models trained primarily on animal data [6].
Applications and Biological Insights
Downstream Task Applications
Single-cell foundation models pretrained using the "cells as sentences" analogy can be adapted to numerous downstream biological tasks through fine-tuning or zero-shot learning. Key applications include:
Cell Type Annotation and Discovery: scFMs can annotate cell types in new datasets based on learned representations, with demonstrated capability for identifying novel cell populations not present in training data [3]. For example, models like scBERT have been specifically designed for cell type annotation tasks [1].
Batch Integration and Data Harmonization: Foundation models effectively remove technical batch effects while preserving biological variation, enabling integration of datasets across different platforms, laboratories, and experimental conditions [18]. This is particularly valuable for large-scale atlas projects combining data from multiple sources.
Gene Function and Regulatory Inference: The attention mechanisms in scFMs can reveal gene-gene relationships and potential regulatory interactions, providing insights into gene function beyond what is available in existing annotations [3]. Analysis of attention weights has shown correspondence with known biological pathways.
Perturbation Prediction and Drug Response Modeling: scFMs can predict cellular responses to genetic perturbations or drug treatments, with applications in pharmaceutical development and personalized medicine [3]. Models have been benchmarked on drug sensitivity prediction across multiple cancer types [3].
Cross-Modal and Cross-Species Transfer Learning: The representations learned by scFMs can transfer across related domains, enabling applications such as protein expression prediction from transcriptomic data or knowledge transfer between model organisms and humans [6].
Biological Interpretation of Model Representations
A critical challenge in scFM development is ensuring that learned representations capture biologically meaningful patterns rather than technical artifacts or spurious correlations. Researchers have developed several approaches to address this challenge:
The scGraph-OntoRWR metric measures the consistency of cell type relationships captured by scFMs with prior biological knowledge encoded in cell ontologies [3]. This provides a biologically grounded evaluation that goes beyond traditional clustering metrics. Similarly, the Lowest Common Ancestor Distance (LCAD) metric assesses the ontological proximity between misclassified cell types, providing a more nuanced evaluation of annotation errors [3].
Analysis of variability in model representations has also proven biologically informative. Studies have revealed that certain neurodevelopmental conditions, including trisomy 21 and CHD8 haploinsufficiency, drive increased gene expression variability in brain cell types [19]. This variability, which is uncoupled from changes in mean transcript abundance, may contribute to the diverse phenotypic outcomes observed in these conditions [19].
Future Directions and Challenges
As single-cell foundation models continue to evolve, several key challenges and opportunities merit attention. Current limitations include the nonsequential nature of omics data, inconsistencies in data quality, computational intensity of training and fine-tuning, and difficulties in interpreting the biological relevance of latent embeddings [1]. Future developments will likely focus on several key areas:
Multimodal Integration: Combining transcriptomic data with other modalities such as epigenomics, proteomics, and spatial information to create more comprehensive foundation models [1] [6]. Techniques like cross-modal graph contrastive learning that combine cellular images with transcriptomic data show particular promise [6].
Interpretability and Explainability: Developing methods to better interpret model predictions and attention mechanisms in biological terms, potentially revealing novel biological insights [1] [3]. This includes refining metrics like scGraph-OntoRWR that evaluate biological consistency.
Efficiency and Scalability: Creating more computationally efficient architectures and training approaches to handle the exponentially growing volumes of single-cell data [1]. This is particularly important as datasets approach billions of cells.
Specialized Domain Models: Developing foundation models tailored to specific biological domains, similar to scPlantLLM for plant genomics [6], but extending to other specialized areas such as immunology, neurobiology, or cancer research.
The analogy of "cells as sentences and genes as words" has provided a powerful framework for applying advanced AI techniques to single-cell biology. As this field matures, scFMs are poised to become indispensable tools for extracting meaningful biological insights from the increasingly complex and high-dimensional data generated by modern single-cell technologies.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling high-resolution analysis of gene expression at the individual cell level, uncovering cellular heterogeneity, developmental trajectories, and complex regulatory networks that were previously obscured by bulk sequencing approaches [6]. However, this transformative technology introduces significant analytical challenges characterized by a trilemma of data quality issues: high sparsity, technical noise, and batch effects. The data are inherently sparse, with a high percentage of zero counts due to both biological absence of expression and technical dropout events [3] [20]. Technical noise arises from amplification biases, stochastic sampling, and other experimental artifacts [21]. Meanwhile, batch effects—technical variations between experiments conducted at different times, locations, or protocols—can confound biological interpretations and impede data integration [21].
The emergence of single-cell foundation models (scFMs) represents a paradigm shift in addressing these challenges. These large-scale artificial intelligence models, pretrained on vast datasets comprising millions of cells, leverage self-supervised learning to capture universal patterns of cellular behavior [1] [3]. Inspired by transformer architectures that revolutionized natural language processing, scFMs learn fundamental biological principles that can be transferred to various downstream tasks through fine-tuning or zero-shot learning [1] [22]. This technical guide examines how scFMs are overcoming persistent data challenges in scRNA-seq analysis, providing researchers with powerful new tools for extracting biological insights from complex single-cell datasets.
Foundation Model Architecture: Biological Language Processing
Conceptual Framework: Cells as Sentences
Single-cell foundation models operate on a powerful analogy: treating individual cells as "sentences" and genes or genomic features as "words" or "tokens" [1] [22]. This conceptual framework allows researchers to apply sophisticated transformer-based architectures originally developed for natural language processing to biological data. By training on massive collections of single-cell transcriptomes encompassing diverse tissues, species, and conditions, these models learn the fundamental "language" of cellular biology [1].
The underlying premise is that exposure to millions of cells across varied biological contexts enables the model to internalize the fundamental principles governing cellular states and functions. This learned knowledge becomes transferable to new datasets and analytical tasks through mechanisms like fine-tuning and zero-shot inference [1] [3]. The model develops rich internal representations that capture biological relationships between genes and cell types, creating a foundation for diverse applications from cell type annotation to perturbation prediction.
Tokenization Strategies: From Expression Values to Model Input
A critical technical challenge in adapting transformer architectures to single-cell data is tokenization—the process of converting raw gene expression profiles into discrete units that the model can process. Unlike words in a sentence, genes have no inherent ordering, requiring strategic approaches to impose structure on the data. Current scFMs employ several tokenization strategies:
- Expression-based ordering: Ranking genes within each cell by their expression levels and feeding the ordered list of top genes as a "sentence" [1]
- Binning approaches: Partitioning genes into bins based on expression values and using these rankings to determine positional encoding [1]
- Normalized counts: Some models report no clear advantage to complex ranking strategies and simply use normalized counts without sophisticated ordering [1]
Each gene is typically represented as a token embedding that combines a gene identifier with its expression value in the given cell. Special tokens may be added to represent cell identity, metadata, or experimental batch information, enabling the model to learn contextual relationships [1]. Positional encoding schemes are adapted to represent the relative order or rank of each gene within the cell's pseudo-sequence.
Transformer Architectures for Single-Cell Data
Most scFMs are built on transformer architectures characterized by attention mechanisms that learn and weight relationships between any pair of input tokens [1]. In practice, this allows the model to determine which genes in a cell are most informative about cellular identity or state, and how they co-vary across cells and conditions. The two primary architectural approaches are:
- Encoder-based models (e.g., BERT-like): Employ bidirectional attention mechanisms that learn from all genes in a cell simultaneously, ideal for classification and embedding tasks [1]
- Decoder-based models (e.g., GPT-like): Use unidirectional masked self-attention that iteratively predicts masked genes conditioned on known genes, particularly effective for generation tasks [1]
The attention layers gradually build latent representations of each cell and gene, capturing hierarchical biological relationships that prove valuable for downstream analytical tasks.
Figure 1: Single-Cell Foundation Model Architecture. scFMs transform raw scRNA-seq data through tokenization strategies and transformer architectures to produce latent representations enabling diverse downstream tasks.
Quantitative Performance Benchmarking
Comparative Performance Across Analytical Tasks
Rigorous benchmarking studies have evaluated scFMs against traditional methods across diverse analytical tasks. A comprehensive 2025 benchmark study evaluated six prominent scFMs (Geneformer, scGPT, UCE, scFoundation, LangCell, and scCello) against established baselines using 12 metrics spanning unsupervised, supervised, and knowledge-based approaches [3]. The results demonstrate that while scFMs are robust and versatile tools, no single model consistently outperforms others across all tasks, emphasizing the importance of task-specific model selection.
Table 1: Performance Comparison of Single-Cell Foundation Models Across Key Tasks
| Model | Batch Integration | Cell Type Annotation | Gene Function Prediction | Perturbation Modeling | Computational Efficiency |
|---|---|---|---|---|---|
| scGPT | High | High | High | High | Medium |
| Geneformer | Medium | High | High | Medium | Medium |
| scFoundation | Medium | Medium | High | Medium | Low |
| scBERT | Low | Medium | Low | Low | High |
| Traditional Methods | Variable | Variable | Low | Low | High |
The benchmarking revealed that scGPT demonstrates robust performance across all tasks, including zero-shot learning and fine-tuning scenarios, while Geneformer and scFoundation show strong capabilities in gene-level tasks [23]. Simpler machine learning models sometimes outperform foundation models in tasks with limited data or when efficiently adapting to specific datasets, particularly under resource constraints [3].
Performance Under Technical Challenges
The ability of scFMs to handle data sparsity and batch effects has been systematically evaluated against traditional methods. A landmark 2023 benchmarking study assessed 46 workflows for single-cell differential expression analysis, examining how batch effects, sequencing depth, and data sparsity impact performance [21]. The findings indicate that:
- For highly sparse data (zero rate > 80%), the use of batch-corrected data rarely improves differential expression analysis
- With substantial batch effects, batch covariate modeling improves analysis compared to naive approaches
- For low-depth data, single-cell techniques based on zero-inflation models deteriorate in performance, whereas analysis of uncorrected data using limmatrend, Wilcoxon test, and fixed effects model performs well [21]
Notably, scFMs have demonstrated particular strength in zero-shot learning scenarios, maintaining high accuracy in cell type annotation and batch integration even on previously unseen data from different species [6]. This capability is particularly valuable for plant single-cell genomics, where models like scPlantLLM overcome issues with batch effect correction and cross-platform data integration that plague traditional methods [6].
Table 2: Method Performance Under Technical Challenges in scRNA-seq Analysis
| Challenge Type | High-Performance Methods | Performance Limitations | Key Considerations |
|---|---|---|---|
| High Sparsity (Zero rate > 80%) | limmatrend, LogN_FEM, DESeq2 | BEC methods show minimal improvement | Avoid zero-inflation models for very sparse data |
| Substantial Batch Effects | MASTCov, ZWedgeR_Cov | Pseudobulk methods perform poorly | Covariate modeling outperforms BEC data |
| Low Sequencing Depth | Wilcoxon test, FEM | scVI+limmatrend effectiveness diminished | Simple methods maintain robustness |
| Cross-Species | scPlantLLM, Geneformer | Animal-trained models on plant data | Species-specific training beneficial |
Experimental Protocols for scFM Implementation
Standardized Framework for Model Application
The application and evaluation of scFMs present significant challenges due to heterogeneous architectures and coding standards. To address this, the BioLLM framework provides a unified interface for integrating and applying diverse scFMs to single-cell RNA sequencing analysis [23]. This standardized approach includes:
- Unified APIs: Standardized application programming interfaces that eliminate architectural and coding inconsistencies
- Comprehensive documentation: Support for streamlined model switching and consistent benchmarking
- Zero-shot and fine-tuning support: Flexible adaptation to various analytical scenarios and resource constraints
The implementation protocol begins with data preprocessing and quality control, followed by model selection based on task requirements and data characteristics, then proceeds to either zero-shot inference or model fine-tuning, and concludes with output interpretation and biological validation [23].
Data Preprocessing and Quality Control
Effective application of scFMs requires careful data preprocessing to ensure model compatibility and reliability. Key steps include:
- Data selection and filtering: Careful selection of datasets, filtering of cells and genes, balancing dataset compositions, and quality controls [1]
- Normalization: Standardization of expression values to mitigate technical variations while preserving biological signals [21]
- Gene selection: Identification of highly variable genes to reduce dimensionality and computational requirements [3]
Assembling high-quality, non-redundant datasets for analysis is as important as model selection for obtaining robust biological insights [1]. For optimal performance, researchers should prioritize data quality over quantity, though scFMs benefit from larger datasets during pretraining.
Model Selection and Fine-Tuning Strategies
Selecting the appropriate scFM requires consideration of multiple factors, including dataset size, task complexity, need for biological interpretability, and available computational resources [3]. Practical guidelines include:
- For large, diverse datasets requiring multiple analytical tasks: scGPT or Geneformer
- For gene-level tasks and network analysis: Geneformer or scFoundation
- For resource-constrained environments or focused tasks: Traditional methods may be preferable
- For plant single-cell data: scPlantLLM specifically designed for plant genomics [6]
Fine-tuning strategies should be tailored to the specific analytical task. For cell type annotation, full fine-tuning on labeled datasets typically yields best results. For batch integration, lighter fine-tuning that preserves the model's general biological knowledge is often more effective [3].
Table 3: Key Research Reagent Solutions for Single-Cell Foundation Model Research
| Resource Category | Specific Examples | Function and Application | Key Features |
|---|---|---|---|
| Data Repositories | CZ CELLxGENE, Human Cell Atlas, GEO/SRA | Provide standardized single-cell datasets for model training and validation | Over 100 million unique cells; standardized annotations [1] |
| Pretrained Models | scGPT, Geneformer, scFoundation, scPlantLLM | Ready-to-use foundation models for various single-cell analysis tasks | Different architectural strengths; species specializations [1] [6] |
| Analysis Frameworks | BioLLM, Seurat, Scanpy | Standardized environments for applying and evaluating scFMs | Unified APIs; benchmarking capabilities [23] |
| Evaluation Metrics | scGraph-OntoRWR, LCAD, ROGI | Specialized metrics for assessing biological relevance of model outputs | Cell ontology-informed evaluation [3] |
| Computational Infrastructure | GPU clusters, Cloud computing platforms | Enable training and deployment of resource-intensive foundation models | Essential for large-scale model training and inference |
Future Directions and Implementation Workflow
Emerging Trends and Development Priorities
The field of single-cell foundation models is rapidly evolving, with several promising directions emerging. Future development priorities include enhancing model interpretability to extract biologically meaningful insights from latent representations, improving scalability to handle increasingly large datasets, and developing better methods for multimodal data integration [1] [6]. Additionally, there is growing interest in incorporating spatial transcriptomics data and single-cell epigenomics to create more comprehensive models of cellular function and regulation [6].
The integration of cross-modal learning approaches, such as graph contrastive learning that combines cellular images with transcriptomic data, shows particular promise for bridging structural and functional genomics [6]. These advancements will not only enrich our knowledge of basic biological processes but also drive innovations in drug development and precision medicine.
Integrated Workflow for Overcoming scRNA-seq Data Challenges
A systematic approach to implementing scFMs for overcoming data challenges involves multiple stages, from data preparation through biological interpretation.
Figure 2: Implementation Workflow for Addressing scRNA-seq Data Challenges. A systematic approach for applying scFMs to overcome sparsity, noise, and batch effects in single-cell data analysis.
The workflow begins with comprehensive data preparation and quality control, followed by assessment of the primary data challenges present in the specific dataset. Based on this assessment, researchers select appropriate models and implementation strategies, such as zero-shot inference for well-represented cell types or fine-tuning for novel cellular states. Finally, biological validation using ontology-informed metrics ensures that computational improvements translate to meaningful biological insights [3].
Single-cell foundation models represent a transformative approach to overcoming the persistent challenges of data sparsity, technical noise, and batch effects in scRNA-seq analysis. By leveraging large-scale pretraining on diverse cellular contexts, these models capture fundamental biological principles that enable robust performance across diverse analytical tasks. While implementation requires careful consideration of model selection, data preparation, and validation strategies, scFMs offer powerful new capabilities for extracting biological insights from complex single-cell datasets. As the field continues to evolve, these models are poised to become indispensable tools for advancing our understanding of cellular biology and driving innovations in drug development and precision medicine.
Inside the Engine Room: Tokenization, Training, and Practical Applications
Single-cell foundation models (scFMs) represent a transformative approach in computational biology, leveraging large-scale deep learning to interpret complex single-cell genomics data. These models are trained on vast datasets encompassing millions of cells to learn fundamental biological principles that generalize across diverse tissues and conditions [1]. The core challenge enabling this technology lies in effectively converting raw gene expression profiles into structured representations that deep learning architectures can process—a procedure known as tokenization.
In natural language processing (NLP), tokenization converts raw text into discrete units called tokens. Similarly, for single-cell data, tokenization transforms gene expression information into a structured format that scFMs can understand and process [1]. This process is foundational because it determines how biological information is encoded and what patterns the model can potentially learn. The tokenization strategy directly impacts a model's ability to capture biological relationships, handle technical variations, and perform accurately on downstream tasks such as cell type annotation, perturbation prediction, and batch integration [3].
Core Components of Tokenization in scFMs
Fundamental Concepts and Definitions
Tokenization in single-cell genomics serves as the critical bridge between biological measurements and computational analysis. Unlike natural language, where words naturally form discrete units, gene expression data presents unique challenges: the data is inherently non-sequential, with no inherent ordering of genes, and exhibits characteristics of high dimensionality and sparsity [1] [3]. The primary goal of tokenization is to overcome these challenges by creating a standardized, structured representation that preserves biological signal while enabling efficient model training.
In practice, tokenization for scFMs involves defining what constitutes a "token" from single-cell data, typically representing each gene or genomic feature as a token. These tokens serve as the fundamental input units for the model, analogous to words in a sentence [1]. The process must also address how to incorporate additional information such as expression values, positional context, and metadata to create rich, informative representations.
Three Core Components of scFM Tokenization
Gene Embeddings
Gene embeddings function as unique identifier vectors for each gene, analogous to word embeddings in NLP. These embeddings allow the model to learn contextual relationships between genes across different cellular environments [3]. Through training on massive single-cell datasets, the model develops embedding spaces where functionally related genes (e.g., those participating in the same biological pathways) are positioned in proximity within the latent space [24]. For example, research has demonstrated that these embeddings can capture protein domain information, gene-disease associations, and transcription factor targets despite being trained solely on expression data [24].
Value Embeddings
Value embeddings represent the expression level of each gene in a specific cell, capturing quantitative information beyond mere gene identity. This component is crucial because the same gene can have dramatically different expression patterns across cell types, states, and conditions [3]. Different strategies exist for handling expression values, including binning approaches that discretize continuous expression values into categorical ranges, and normalized count representations that maintain relative expression levels [1] [24]. The "Binning-By-Gene" method has shown particular promise by allocating gene expressions across samples into bins based on expression rank, reducing bias toward genes with atypical expression distributions [24].
Positional Embeddings
Positional embeddings address the non-sequential nature of genomic data by providing artificial ordering information to the model. Since genes lack inherent sequence in expression data, various strategies have emerged for imposing structure, including expression-based ordering (ranking genes by expression level within each cell) and genomic coordinate-based ordering [1] [3]. These embeddings enable the transformer architecture to recognize relationships between genes regardless of their arbitrary position in the input sequence. Some models employ Attention with Linear Biases (ALiBi) as an alternative to traditional positional embeddings, particularly beneficial for handling long input sequences [25].
Table 1: Core Components of Tokenization in Single-Cell Foundation Models
| Component | Function | Implementation Examples | Biological Significance |
|---|---|---|---|
| Gene Embeddings | Unique identifier for each gene | Learned vectors for each gene identifier | Captures functional relationships between genes across cellular contexts |
| Value Embeddings | Represents expression magnitude | Binned expression values; Normalized counts | Encodes quantitative gene activity levels essential for understanding cell state |
| Positional Embeddings | Provides artificial sequence context | Expression-based ordering; Genomic coordinates; ALiBi | Enables attention mechanisms to model gene-gene interactions despite non-sequential nature |
Tokenization Strategies and Architectures
Input Representation Strategies
The conversion of raw gene expression data into tokenized inputs requires careful consideration of biological principles and computational efficiency. A key challenge is that gene expression data lacks natural ordering, unlike words in a sentence [1]. To address this, several strategic approaches have been developed:
Expression-based ordering involves ranking genes within each cell by their expression levels, feeding the ordered list of top genes as a "sentence" to the model [1] [24]. This provides a deterministic sequence based on expression magnitude, creating a consistent input structure while prioritizing highly expressed genes that may be most biologically relevant. For example, the GeneRAIN model sorts genes from highest to lowest based on expression z-scores before processing [24].
Partitioning approaches involve binning genes by their expression values and using these rankings to determine positional encoding [1]. This method can reduce noise from small expression variations while maintaining the relative abundance information crucial for understanding cellular state. Some implementations combine partitioning with specialized normalization techniques like the "Binning-By-Gene" method, which allocates gene expressions across samples into bins based on expression rank, ensuring equal probability for each gene to occupy any rank position [24].
Multi-modal tokenization incorporates additional data types beyond gene expression. Advanced scFMs can include tokens representing different sequencing modalities (e.g., scATAC-seq for chromatin accessibility), spatial context, or protein abundance measurements [1] [8]. Special modality tokens are often prepended to indicate the data type, enabling the model to learn cross-modal relationships and integrate complementary biological information.
Model Architecture Considerations
Tokenization strategies are intimately connected to model architecture decisions, with different architectural families presenting distinct advantages for single-cell data:
Encoder-based models (e.g., BERT-style) use bidirectional attention mechanisms that learn from all genes in a cell simultaneously [1]. These architectures are particularly effective for classification tasks like cell type annotation and embedding generation. The bidirectional nature allows the model to capture complex, interdependent relationships between genes, mirroring the biological reality of coordinated gene regulation networks.
Decoder-based models (e.g., GPT-style) employ unidirectional masked self-attention that iteratively predicts masked genes conditioned on known genes [1] [24]. These excel at generative tasks and sequence completion, potentially offering advantages for predicting cellular responses to perturbations or generating synthetic expression profiles. The GeneRAIN project found that GPT-style architectures trained to predict the next gene in an expression-ordered sequence effectively captured biological relationships [24].
Hybrid and specialized architectures continue to emerge, combining elements of both approaches or introducing novel mechanisms to address specific biological challenges. For example, Nicheformer integrates single-cell analysis with spatial transcriptomics, requiring specialized tokenization strategies that preserve spatial context [8]. Similarly, mRNABERT implements a dual tokenization scheme that treats individual nucleotides as tokens for untranslated regions (UTRs) and codons for coding sequences (CDS), aligning tokenization with biological structure [25].
Table 2: Comparison of Tokenization Approaches Across Model Architectures
| Architecture Type | Tokenization Strategy | Advantages | Common Applications |
|---|---|---|---|
| Encoder-based (BERT) | Whole-cell masking with bidirectional context | Captures complex gene interactions; Strong representation learning | Cell type annotation; Batch integration; Gene function prediction |
| Decoder-based (GPT) | Sequential prediction with causal masking | Effective for generation; Can simulate trajectories | Perturbation response prediction; Synthetic data generation |
| Hybrid Architectures | Multi-modal tokens; Custom ordering schemes | Flexibility for specialized tasks; Integration of diverse data types | Spatial transcriptomics; Multi-omics integration; Therapeutic design |
Experimental Protocols and Implementation
Detailed Methodologies for Tokenization
Implementing effective tokenization requires careful attention to both biological principles and computational practicalities. Below are detailed protocols derived from successful implementations:
Protocol 1: Expression-Based Tokenization with Binning Normalization
This protocol is adapted from the GeneRAIN implementation, which demonstrated superior performance in learning gene biological attributes [24]:
Data Preprocessing: Begin with raw count matrix of cells × genes. Normalize each sample by library size to a total count of 10 million using count per million (CPM) or similar approach.
Binning-by-Gene Normalization: For each gene across all samples, allocate expressions into 2,000 bins based on expression rank. Genes with zero expression are allocated to the lowest bin, while non-zero expressions are evenly distributed across remaining bins.
Input Sequence Construction: For each cell, sort genes from highest to lowest based on their binned expression values. Select the top N genes (typically 1,000-2,000) based on model capacity.
Token Creation: For each selected gene, create a composite token embedding that combines:
- Gene identity embedding (learned)
- Expression value embedding (based on bin index)
- Positional embedding (based on rank in sorted sequence)
Model Input: Feed the sequence of composite tokens into the transformer architecture for self-supervised pretraining.
Protocol 2: Dual Tokenization for Sequence-Specific Regions
For applications involving specific genomic sequences rather than expression profiles, such as mRNA design, mRNABERT implements a specialized dual tokenization approach [25]:
Region Identification: Segment full-length mRNA sequences into distinct regions: 5' UTR, coding sequence (CDS), and 3' UTR.
Region-Specific Tokenization:
- For UTR regions: Tokenize at nucleotide level (A, C, G, U as tokens)
- For CDS regions: Tokenize at codon level (three-nucleotide units as tokens)
Sequence Integration: Combine region-specific tokens into a unified sequence, inserting special tokens to indicate region boundaries.
Positional Encoding: Implement Attention with Linear Biases (ALiBi) to handle long sequences and provide positional context without traditional positional embeddings.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for Tokenization Implementation
| Resource Type | Specific Examples | Function in Tokenization | Availability |
|---|---|---|---|
| Data Repositories | CZ CELLxGENE; Human Cell Atlas; PanglaoDB | Provide standardized single-cell datasets for pretraining | Publicly available |
| Processing Tools | Seurat; Scanpy; scvi-tools | Preprocessing and normalization of raw scRNA-seq data | Open source |
| Tokenization Libraries | Hugging Face Tokenizers; SentencePiece | Implement BPE, WordPiece, and custom tokenization algorithms | Open source |
| Model Frameworks | PyTorch; TensorFlow; JAX | Enable custom model architecture implementation | Open source |
| Benchmarking Suites | scGraph-OntoRWR; Attribute Learning Index | Evaluate biological relevance of learned representations | Research implementations |
Advanced Tokenization Applications
Multi-Modal and Spatial Tokenization
As single-cell technologies evolve beyond transcriptomics to capture multi-dimensional cellular characteristics, tokenization strategies have expanded accordingly. Spatial tokenization represents a particularly advanced approach that integrates physical location context with molecular measurements. Nicheformer, the first large-scale foundation model for single-cell and spatial omics, demonstrates this capability by learning from both dissociated single-cell data and spatial transcriptomics [8]. The model tokenizes spatial coordinates alongside gene expression values, enabling it to reconstruct tissue organization patterns and model cell-cell interactions that would be lost in conventional single-cell analysis.
Multi-modal tokenization incorporates diverse data types such as chromatin accessibility (scATAC-seq), protein abundance (CITE-seq), and genetic variants. This approach uses special modality tokens to indicate data type and often employs cross-modal attention mechanisms that allow information to flow between different measurement types [1]. For example, a single model might process both gene expression tokens and chromatin accessibility tokens, learning the complex relationships between epigenetic state and transcriptional output.
Biological Sequence Tokenization
Beyond gene expression profiles, tokenization strategies have been adapted for biological sequences including DNA, RNA, and protein sequences. mRNABERT introduces a dual tokenization scheme that handles different regions of mRNA molecules with appropriate granularity: nucleotide-level tokenization for untranslated regions (UTRs) and codon-level tokenization for coding sequences (CDS) [25]. This biologically-informed approach respects the different functional constraints acting on various mRNA regions, resulting in improved performance for therapeutic mRNA design tasks.
For genomic sequences, traditional approaches often used single-nucleotide or k-mer tokenization, but recent advances employ data-driven tokenization methods like Byte-Pair Encoding (BPE) adapted from NLP [26] [27]. These methods can identify biologically meaningful units in DNA sequences, such as conserved motifs or regulatory elements, and tokenize them as single units, improving model efficiency and interpretability.
Evaluation and Benchmarking
Metrics for Tokenization Quality Assessment
Evaluating the effectiveness of tokenization strategies requires specialized metrics that capture both computational efficiency and biological relevance:
The Attribute Learning Index is a comprehensive metric that evaluates how well gene embeddings capture biological attributes [24]. It averages three clustering consistency metrics (Adjusted Rand Index, Fowlkes-Mallows Index, and Normalized Mutual Information) between model embedding-based clustering and actual gene biological attribute groupings. This index provides a quantitative measure of the biological information encoded in the token representations.
scGraph-OntoRWR is a novel metric designed specifically for scFMs that measures the consistency of cell type relationships captured by the model with prior biological knowledge [3]. By comparing the relational structure of cell types in the embedding space with established biological hierarchies, this metric evaluates whether the tokenization and subsequent representation learning capture meaningful biological relationships beyond superficial patterns.
Lowest Common Ancestor Distance (LCAD) measures the ontological proximity between misclassified cell types, assessing the severity of errors in cell type annotation tasks [3]. This biologically-informed metric recognizes that misclassifying between closely related cell types (e.g., two T cell subtypes) is less severe than misclassifying between distantly related types (e.g., neuron vs. immune cell), providing a more nuanced evaluation of model performance.
Performance Comparison
Benchmarking studies reveal that tokenization strategies significantly impact model performance across diverse biological tasks. Research comparing six prominent scFMs against traditional methods found that while foundation models generally offer robustness and versatility, no single approach consistently outperforms others across all tasks [3]. This underscores the importance of selecting tokenization strategies aligned with specific application requirements.
The GeneRAIN project demonstrated that their Binning-By-Gene normalization method significantly enhanced model capability in learning gene biological attributes compared to z-score based approaches (p = 0.007 by t-test) [24]. Similarly, mRNABERT's dual tokenization scheme outperformed previous models in the majority of tasks for 5' UTR and CDS design, RNA-binding protein site prediction, and full-length mRNA property prediction [25].
Future Directions and Challenges
The development of tokenization strategies for single-cell foundation models faces several important challenges and opportunities for advancement. A primary limitation is the non-sequential nature of omics data, which requires artificial ordering schemes that may not fully capture biological relationships [1]. Future approaches may explore graph-based representations that more naturally model gene regulatory networks, with tokenization adapted to handle graph structures rather than linear sequences.
Interpretability remains a significant challenge, as understanding what biological information is captured in token embeddings and model representations remains nontrivial [1]. Research into explainable AI methods tailored to tokenization could help bridge this gap, potentially leading to novel biological insights discovered through model interpretation rather than confirmation of known biology.
Computational efficiency continues to constrain tokenization approaches, particularly as datasets grow to hundreds of millions of cells [1]. Emerging architectures like state space models (e.g., Mamba) and efficient attention mechanisms may enable more scalable tokenization while maintaining biological fidelity [27]. Additionally, specialized tokenization for long biological sequences represents an active area of innovation, with methods like HyenaDNA demonstrating capabilities for processing sequences up to 1 million base pairs [27].
As single-cell technologies continue to evolve, tokenization strategies must adapt to incorporate new data modalities, spatial contexts, and temporal dynamics. The ultimate goal remains the development of representations that faithfully capture biological reality while enabling powerful deep learning models to extract meaningful patterns and relationships. Through continued refinement of tokenization approaches, single-cell foundation models will advance our understanding of cellular function and drive innovations in therapeutic development.
Single-cell RNA sequencing (scRNA-seq) generates high-dimensional data that captures the transcriptomic state of individual cells, but this data lacks an inherent sequential order. This paper explores the critical role of positional encoding, a technique adapted from transformer-based natural language processing, in enabling single-cell foundation models (scFMs) to understand and interpret the complex, non-sequential relationships within genomic data. We provide a detailed technical examination of how various positional encoding strategies—including sinusoidal, learnable, and rank-value encoding—are implemented to inject positional information into scFMs, thereby allowing them to capture cellular heterogeneity, gene-gene relationships, and spatial context. Supported by quantitative comparisons and detailed experimental protocols from state-of-the-art models, this guide serves as a comprehensive resource for researchers and drug development professionals aiming to leverage foundation models for advanced genomic analysis.
In natural language processing, the meaning of a sentence changes fundamentally with the order of its words (e.g., "Allen walks dog" vs. "dog walks Allen") [28]. Similarly, in genomics, the sequence of genes and their expression levels defines cellular identity and function. However, unlike language, which has a natural left-to-right sequence, genomic data from technologies like scRNA-seq is inherently non-sequential; it consists of unordered sets of gene expression values for each cell.
Transformers, the architecture underlying modern foundation models, process all elements of their input in parallel and lack any inherent mechanism to understand order [29] [30]. Positional encoding solves this by explicitly injecting information about position or order into the model, a technique that is equally vital for single-cell data as it is for language [2]. This allows the model to distinguish not only what genes are expressed but also to learn from how they are ordered or positioned relative to one another, whether in a ranked list or a spatial context.
The Mechanics of Positional Encoding in Transformers
Core Mathematical Formulation
The most widely recognized method, introduced in the "Attention Is All You Need" paper, uses sinusoidal functions to create a unique, deterministic encoding for each position in a sequence [29] [28]. For a model with an embedding dimension of d_model, the positional encoding (PE) for a word at position pos is a vector where each element is calculated as follows [29] [31] [28]:
- Even indices: ( PE{(pos, 2i)} = \sin\left(\frac{pos}{n^{2i/d{\text{model}}}}\right) )
- Odd indices: ( PE{(pos, 2i+1)} = \cos\left(\frac{pos}{n^{2i/d{\text{model}}}}\right) )
Here, i is the dimension index (from 0 to d_model/2 - 1), and n is a user-defined scalar, typically 10,000 [29]. This scheme ensures each position receives a unique encoding and that the model can learn relative positions due to the trigonometric properties of the functions [30].
Key Properties and Advantages
Sinusoidal positional encoding possesses several properties that make it particularly effective [29] [30]:
- Normalization: Values are bounded between -1 and 1, preventing numerical instability.
- Uniqueness: Each position in the sequence is assigned a unique representation.
- Relative Position Awareness: The encoding for a position offset by a fixed value
kcan be represented as a linear function of the original encoding, making it easier for the model to learn and attend to relative positions.
Table 1: Advantages and Limitations of Sinusoidal Positional Encoding
| Advantage | Technical Rationale | Limitation | |
|---|---|---|---|
| Deterministic & Fixed | No learned parameters; generalizes to sequences of unseen lengths during training [32]. | Fixed Length Limit | The original approach struggles with sequences longer than those seen in training [31] [33]. |
| Extrapolation | Smooth, periodic nature allows the model to handle longer sequences [32]. | Static Patterns | Cannot adapt position patterns based on specific data or tasks [32]. |
| Relative Positioning | ( PE(pos + k) ) can be derived as a linear function of ( PE(pos) ) [30]. | Absolute Focus | Primarily encodes absolute position; relative positioning must be learned [33]. |
Positional Encoding Strategies for Single-Cell Foundation Models
Single-cell foundation models adapt the core principle of positional encoding to represent the "position" of a gene within the context of a cell's transcriptome. The "sequence" can be artificially constructed through various tokenization strategies.
Table 2: Positional Encoding and Tokenization in Single-Cell Foundation Models
| Tokenization Strategy | Positional Encoding Method | Representative Model(s) | Key Advantage |
|---|---|---|---|
| Gene Ranking | The position in a sequence of genes ordered by expression level. | Geneformer [34], scGPT [34], Nicheformer [10] | Robust to batch effects; preserves gene-gene relationships [10]. |
| Value Categorization | The position in a sequence of (gene, expression bucket) pairs. | scBert [34] | Converts continuous prediction into a classification problem. |
| Value Projection | The gene's position in the fixed, canonical list of all genes. | scFoundation [34], CellFM [34] | Preserves the full resolution and continuous nature of gene expression data. |
Rank-Based Encoding for Cellular Context
In models like Geneformer and Nicheformer, a cell is represented as a sequence of gene tokens sorted from highest to lowest expression [34] [10]. The position of a gene in this ranked list becomes its positional encoding. This approach, known as rank-value encoding, is akin to asking "where does this word occur in a sentence?" for each gene [2]. For example, a highly expressed gene like ACTB might consistently appear in the first few positions across many cells, providing a strong, context-aware signal to the model.
Experimental Protocol for Rank-Based Pre-training:
- Input: A vector of raw gene expression counts for a single cell.
- Tokenization: Genes are sorted by their expression value, from highest to lowest.
- Sequence Creation: The corresponding gene tokens are arranged in this sorted order to form the input sequence. The model is trained to predict the rank of genes or their relationships within this sequence.
- Objective: The model learns contextual embeddings for genes based on their co-occurrence and relative positions across millions of cells [10].
Absolute and Spatial Encoding for Multi-Modal Integration
More advanced models incorporate absolute positional information. Nicheformer is pre-trained on both dissociated single-cell data and spatially resolved transcriptomics data [10]. It uses contextual tokens to encode the absolute "position" in terms of technology modality (e.g., dissociated vs. MERFISH vs. Xenium) and species (human vs. mouse). This allows the model to learn a joint, spatially aware representation.
PEGTB-MIL, a model designed for histopathology whole-slide images, explicitly encodes the 2D spatial coordinates of tissue patches [35]. It uses a position encoder module to convert normalized (x, y) coordinates into spatial embeddings, which are fused with the patch's semantic features. An auxiliary position decoder module is used during training to reconstruct the original coordinates, ensuring spatial-semantic consistency [35].
Quantitative Analysis of Model Performance
The effectiveness of positional encoding and sophisticated model architecture is demonstrated by the state-of-the-art performance of recent scFMs on diverse downstream tasks.
Table 3: Performance of Single-Cell Foundation Models on Key Tasks
| Model | Pre-training Scale | Key Downstream Task | Reported Performance |
|---|---|---|---|
| CellFM [34] | 100M human cells, 800M parameters | Cell annotation, Perturbation prediction | Outperforms existing models in gene function prediction and capturing gene-gene relationships. |
| Nicheformer [10] | 110M cells (dissociated & spatial) | Spatial composition prediction, Spatial label transfer | Systematically outperforms Geneformer, scGPT, and UCE on spatially-aware tasks. |
| PEGTB-MIL [35] | Two TCGA and two in-house clinical datasets | Lung and breast cancer subtyping, EGFR & KIT mutation prediction | Achieves superior classification performance compared to state-of-the-art MIL-based methods. |
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Components for a Single-Cell Foundation Model Pipeline
| Component / Reagent | Function in the Experimental Workflow |
|---|---|
| scRNA-seq Library (e.g., 10x Genomics 3') | Generates the raw transcriptomic data from individual cells; the primary source of input data [34]. |
| Spatial Transcriptomics (e.g., MERFISH, Xenium) | Provides spatially resolved gene expression data for training models like Nicheformer [10]. |
| Positional Encoding Module | Algorithmic component that injects order or positional information into the model (e.g., rank-based, 2D coordinate) [35] [10]. |
| Multi-Head Self-Attention Layer | The core transformer component that allows the model to learn dependencies between all genes in the context of their positions [35]. |
| Pre-training Corpus (e.g., SpatialCorpus-110M) | A large, curated collection of single-cell datasets used for initial self-supervised learning of the foundation model [10]. |
Positional encoding has evolved from a method to specify word order in sentences to a flexible, powerful tool for imparting structural meaning to non-sequential genomic data. By enabling single-cell foundation models to understand context through gene ranking, spatial coordinates, or other metadata, it forms the bedrock upon which these models learn complex biological relationships. As evidenced by the performance of models like CellFM, Nicheformer, and PEGTB-MIL, the strategic application of positional encoding is pivotal for tasks ranging from basic cell annotation to predicting spatial organization and gene mutation status. This capability is fundamental for accelerating drug discovery and deepening our understanding of cellular biology.
Masked gene prediction has emerged as a foundational self-supervised learning paradigm for single-cell genomics, enabling models to learn rich biological representations by reconstructing randomly obscured portions of gene expression data. This technical guide examines the architectural principles, methodological variations, and experimental protocols underlying this pretraining approach, which forms the core of modern single-cell foundation models (scFMs). We detail how models trained to predict masked genes develop a profound understanding of gene-gene relationships and cellular states, facilitating transfer learning across diverse downstream applications from cell type annotation to perturbation response prediction. Within the broader thesis of single-cell foundation model research, masked gene prediction represents a pivotal methodological advancement that leverages the inherent structure of transcriptomic data without requiring expensive labeled datasets, thereby accelerating discoveries in basic biology and therapeutic development.
The exponential growth of single-cell RNA sequencing (scRNA-seq) data has created unprecedented opportunities for understanding cellular heterogeneity at transcriptomic resolution. However, the characteristic high dimensionality, technical noise, and sparsity of these datasets present significant analytical challenges [1] [3]. Masked gene prediction addresses these challenges through a self-supervised pretraining objective where models learn to reconstruct randomly masked portions of gene expression profiles based on the remaining observable context [1] [36]. This approach enables scFMs to capture the complex, context-dependent relationships between genes that define cellular identity and function.
Inspired by masked language modeling in natural language processing (NLP), where models predict missing words in sentences, masked gene prediction treats cells as "sentences" and genes as "words" [1] [37]. By training on massive datasets comprising millions of cells across diverse tissues and conditions, models learn a fundamental "language of cells" that encodes biological principles transferable to various downstream tasks. This pretraining paradigm has become the cornerstone of leading scFMs including scGPT, scBERT, Geneformer, and scFoundation, establishing masked prediction as a dominant approach in the field [1] [38] [37].
Conceptual Framework and Architectural Foundations
Core Principles of Masked Autoencoding
Masked autoencoding for gene prediction operates on a conceptually simple yet powerful principle: given a partial view of a cell's gene expression profile, the model must infer the missing values based on patterns learned from vast cellular datasets [36]. This self-supervised objective forces the model to develop an understanding of:
- Gene co-expression patterns: Which genes tend to be expressed together across different cellular contexts
- Regulatory relationships: How transcription factors and their targets correlate
- Pathway dependencies: The logical structure of biological pathways and processes
- Cellular states: How gene expression patterns define distinct cell types and states
The training process involves randomly masking a portion (typically 15-30%) of the input genes and training the model to minimize the difference between predicted and actual expression values for these masked elements [36] [38]. Through this process, the model develops a comprehensive understanding of transcriptional regulation without any explicit labeling.
Model Architecture Components
Table 1: Key Architectural Components in Masked Gene Prediction Models
| Component | Function | Common Implementations |
|---|---|---|
| Tokenization | Converts raw gene expression into model inputs | Gene ranking, value binning, direct normalization [1] |
| Embedding Module | Represents genes and values in high-dimensional space | Gene embeddings, value embeddings, positional encodings [1] [38] |
| Backbone Architecture | Processes token sequences to build representations | Transformer encoders, RetNet, specialized attention mechanisms [1] [38] |
| Prediction Head | Reconstructs masked gene values | Linear projection, categorical classification, regression [1] [36] |
| Masking Strategy | Determines which genes to obscure during training | Random masking, gene program masking, adaptive strategies [36] |
Most scFMs utilizing masked gene prediction build upon transformer architectures, which employ self-attention mechanisms to model relationships between all genes in a cell simultaneously [1] [37]. The attention mechanism allows the model to weight the importance of different observable genes when predicting masked ones, effectively learning which genes are most informative for inferring others in specific cellular contexts.
Methodological Approaches and Experimental Protocols
Data Preprocessing and Tokenization Strategies
A critical first step in masked gene prediction is converting continuous, high-dimensional gene expression data into a structured format suitable for model input. The non-sequential nature of genomic data presents a unique challenge compared to natural language, requiring thoughtful tokenization strategies:
Gene Ranking Approach: Genes are ordered by expression level within each cell, creating a deterministic sequence where highly expressed genes appear first [1] [37]. Models like Geneformer use this approach, treating gene ranks as tokens.
Value Binning Strategy: Continuous expression values are discretized into bins or "buckets," converting regression to classification [1]. scBERT employs this method, binning expression values into categories that become input tokens.
Direct Value Projection: Some recent models like scFoundation and CellFM project normalized expression values directly, preserving the full resolution of the data [38].
Additionally, special tokens may be incorporated to represent metadata such as cell type, batch information, or experimental conditions, enabling the model to learn context-dependent relationships [1].
Masking Strategies and Their Biological Rationale
The strategy for selecting which genes to mask during training significantly impacts what biological relationships the model learns. Different approaches include:
Random Masking: The simplest approach where genes are masked uniformly at random, introducing minimal inductive bias [36].
Gene Program Masking: Masking functionally related gene sets (e.g., pathways, co-expression modules) to force the model to learn higher-order regulatory relationships [36].
Adaptive Masking: Strategically selecting genes based on importance metrics or functional annotations to focus learning on biologically significant relationships.
Empirical evidence suggests that more biologically-informed masking strategies, such as gene program masking, can enhance model performance on downstream tasks requiring understanding of functional relationships [36].
Experimental Protocol for Pretraining scFMs
The following protocol outlines the standard procedure for pretraining a single-cell foundation model using masked gene prediction:
Step 1: Data Collection and Curation
- Aggregate single-cell datasets from public repositories (CELLxGENE, GEO, SRA)
- Target diversity in tissues, conditions, and technologies
- Apply quality control filters (minimum genes/cell, maximum mitochondrial percentage)
- Standardize gene annotations using HUGO Gene Nomenclature Committee guidelines
Step 2: Data Preprocessing
- Normalize expression values to account for sequencing depth variations
- Select highly variable genes to focus on biologically informative features
- Apply appropriate transformation (log, square root) to stabilize variance
- Implement tokenization strategy (ranking, binning, or projection)
Step 3: Model Configuration
- Initialize transformer-based architecture with appropriate dimensions
- Set masking ratio (typically 15-30%)
- Configure optimization parameters (learning rate, batch size, warmup steps)
- Implement gradient checkpointing if facing memory constraints
Step 4: Training Procedure
- Iterate through training corpus with random masking applications
- Compute reconstruction loss between predicted and actual expression values
- Update parameters via backpropagation
- Monitor validation loss for convergence and potential overfitting
Step 5: Model Evaluation and Validation
- Assess reconstruction accuracy on held-out validation sets
- Evaluate learned representations via downstream tasks (cell type annotation, batch correction)
- Perform biological validation (pathway enrichment, gene-gene relationship analysis)
Diagram 1: Masked Gene Prediction Workflow
Performance Benchmarks and Quantitative Analysis
Comparative Performance Across Downstream Tasks
Table 2: Performance of Masked Gene Prediction Models on Key Benchmarks
| Model | Training Scale | Cell Annotation (F1) | Perturbation Prediction (AUPRC) | Batch Correction (ASW) | Gene Function (AUROC) |
|---|---|---|---|---|---|
| scGPT [36] [23] | 33M cells | 0.85 | 0.79 | 0.88 | 0.82 |
| Geneformer [3] [23] | 30M cells | 0.81 | 0.76 | 0.85 | 0.85 |
| scBERT [1] [23] | 10M cells | 0.78 | 0.72 | 0.82 | 0.78 |
| scFoundation [3] [38] | 50M cells | 0.83 | 0.81 | 0.86 | 0.84 |
| CellFM [38] | 100M cells | 0.87 | 0.84 | 0.91 | 0.87 |
| UCE [3] | 36M cells | 0.82 | 0.78 | 0.87 | 0.83 |
Empirical evaluations demonstrate that models pretrained with masked gene prediction objectives consistently outperform traditional methods and supervised baselines, particularly in transfer learning scenarios where models are applied to datasets not seen during training [36] [3]. The scale of pretraining data emerges as a critical factor, with models trained on larger and more diverse datasets (e.g., CellFM with 100M cells) generally achieving superior performance across multiple benchmarks [38].
Impact of Pretraining on Downstream Task Performance
Studies systematically evaluating the value of self-supervised pretraining have revealed several key patterns:
- Masked autoencoders consistently outperform contrastive learning methods in single-cell genomics, contrary to trends observed in computer vision [36]
- The benefits of pretraining are most pronounced in transfer learning scenarios, where models leverage knowledge from large auxiliary datasets to analyze smaller target datasets [36]
- Pretraining significantly improves performance on rare cell type identification, with one study reporting macro F1 score improvements from 0.70 to 0.75 in peripheral blood mononuclear cells [36]
- The advantages scale with dataset diversity, with models pretrained on more donors showing greater performance gains [36]
Research Reagent Solutions for scFM Development
Table 3: Essential Resources for Implementing Masked Gene Prediction
| Resource Category | Specific Tools/Platforms | Function and Application |
|---|---|---|
| Data Repositories | CELLxGENE, GEO, SRA, ENA, GSA | Provide standardized access to millions of single-cell datasets for pretraining [1] [38] |
| Preprocessing Tools | Scanpy, Seurat, SynEcoSys | Perform quality control, normalization, and feature selection on raw scRNA-seq data [38] |
| Model Frameworks | PyTorch, TensorFlow, MindSpore | Provide foundational infrastructure for implementing and training transformer architectures [38] |
| Specialized Architectures | Transformer, RetNet, LoRA | Enable efficient attention mechanisms and parameter-efficient fine-tuning [38] |
| Evaluation Suites | BioLLM, scGraph-OntoRWR | Standardize benchmarking across multiple downstream tasks and biological metrics [3] [23] |
The development of effective masked gene prediction models requires both biological data resources and computational infrastructure. Large-scale pretraining typically necessitates high-performance computing environments with multiple GPUs or specialized AI accelerators (e.g., Ascend NPUs) [38]. Frameworks like BioLLM have emerged to standardize model evaluation and comparison, addressing the challenge of heterogeneous architectures and implementation standards across different scFMs [23].
Applications in Biological Research and Drug Development
The representations learned through masked gene prediction pretraining enable a wide range of applications with significant implications for biological discovery and therapeutic development:
Cell Type Annotation and Novel Cell Identification: Pretrained models can accurately assign cell identity labels in new datasets and identify previously uncharacterized cell states based on their transcriptional profiles [3] [39].
Perturbation Response Prediction: By understanding the relationships between genes, models can predict how cells will respond to genetic perturbations or drug treatments, enabling in silico screening of therapeutic candidates [3] [38].
Gene Function and Interaction Inference: The attention mechanisms in transformer models can reveal functional relationships between genes, potentially identifying novel regulatory interactions or pathway memberships [3].
Multi-Omics Integration: Models can incorporate additional data modalities such as ATAC-seq or proteomics by extending the masking approach to multiple feature types, creating unified representations of cellular state [1].
Clinical Application and Biomarker Discovery: The ability to identify subtle transcriptional patterns makes these models valuable for identifying diagnostic and prognostic biomarkers from patient samples [3].
Diagram 2: Research Applications of Pretrained Single-Cell Foundation Models
Future Directions and Methodological Challenges
Despite the considerable success of masked gene prediction in single-cell foundation models, several challenges and opportunities for advancement remain:
Interpretability and Biological Validation: While models demonstrate impressive performance on practical tasks, directly interpreting the biological knowledge encoded in their parameters remains challenging [1] [3]. Developing methods to extract and validate this knowledge is an active research area.
Computational Efficiency: Training large-scale transformer models on millions of cells requires substantial computational resources [1] [38]. Approaches such as efficient attention mechanisms (e.g., RetNet in CellFM) and parameter-efficient fine-tuning help address these constraints [38].
Multimodal Integration: Current models primarily focus on transcriptomic data, but integrating additional modalities (epigenomics, proteomics, spatial context) will create more comprehensive cellular representations [1].
Handling Technical Artifacts: Models must robustly handle batch effects, sequencing platform differences, and other technical variations while preserving biological signals [3] [40].
Rare Cell Type Considerations: Standard pretraining approaches may underrepresent rare cell populations [40]. Developing specialized strategies to ensure these populations are adequately captured represents an important direction for future work.
The continued evolution of masked gene prediction methodologies will further enhance our ability to extract meaningful biological insights from single-cell genomics data, ultimately advancing both basic scientific understanding and therapeutic development.
Single-cell foundation models (scFMs) represent a transformative advancement in computational biology, leveraging large-scale deep learning architectures pre-trained on vast single-cell datasets to achieve remarkable versatility across downstream tasks. These models are defined by their large-scale, self-supervised training on diverse datasets, enabling adaptation to a wide range of biological questions through fine-tuning or zero-shot learning [1] [2]. The emergence of scFMs marks a significant evolution from traditional single-cell analysis pipelines, which typically required specialized tools for each analytical step. Instead, scFMs provide a unified framework capable of addressing diverse challenges—from basic cell type annotation to complex clinical predictions like drug response—using a single, foundational architecture [1] [3].
The conceptual underpinning of scFMs draws inspiration from the success of transformer-based models in natural language processing (NLP). In this analogy, individual cells are treated as "sentences," while genes or genomic features, along with their expression values, serve as "words" or "tokens" [1] [2]. By training on millions of cells encompassing diverse tissues, species, and biological conditions, scFMs learn the fundamental "language" of cellular biology, capturing intricate patterns of gene expression, regulation, and interaction that generalize across experimental contexts [1]. This approach has positioned scFMs as powerful tools for extracting biologically meaningful insights from the rapidly expanding repositories of single-cell genomic data, effectively addressing the critical need for unified analytical frameworks in the field [1].
Architectural Foundations of Single-Cell Foundation Models
Core Model Architecture and Components
Most scFMs are built on transformer architectures, which utilize attention mechanisms to model complex dependencies between input tokens. The self-attention mechanism allows these models to dynamically weight the importance of different genes when making predictions, effectively learning which genes are most informative for specific biological contexts [1]. Two predominant architectural variants have emerged: encoder-based models (e.g., scBERT) that use bidirectional attention to learn from all genes in a cell simultaneously, and decoder-based models (e.g., scGPT) that employ masked self-attention to iteratively predict masked genes conditioned on known genes [1]. Hybrid designs combining both approaches are also being explored, though no single architecture has yet emerged as clearly superior for all single-cell data tasks [1].
The input processing for scFMs involves several critical components that transform raw single-cell data into model-readable inputs. Gene embeddings convert gene identifiers into continuous vector representations, analogous to word embeddings in NLP. Value embeddings encode the expression levels of each gene, often through binning or normalization strategies. Positional embeddings present a unique challenge since gene expression data lacks natural sequential ordering; most models address this by ranking genes by expression levels within each cell to create a deterministic sequence [1]. Additional special tokens may be incorporated to represent cell-level metadata, experimental batch information, or modality indicators for multi-omics applications [1] [2].
Pretraining Strategies and Data Infrastructure
ScFMs are typically pre-trained using self-supervised objectives on massive, aggregated single-cell datasets. Public data archives such as CZ CELLxGENE, which provides standardized access to over 100 million unique cells, along with resources from the Human Cell Atlas, GEO, SRA, and curated compendia like PanglaoDB, form the essential training corpora for these models [1]. The self-supervised pretraining tasks often involve predicting masked portions of the input data, such as randomly masked gene expression values, which forces the model to learn meaningful representations of cellular states based on contextual gene relationships [1].
This pretraining paradigm allows scFMs to develop rich internal representations of biological knowledge that can be transferred to downstream tasks with minimal additional training. The scale and diversity of the pretraining data are critical factors in model performance, as they enable the capture of universal patterns applicable to various biological contexts and experimental conditions [1]. However, challenges in data quality, including batch effects, technical noise, and varying processing steps across studies, necessitate careful data selection and preprocessing to build robust foundation models [1].
Methodological Framework for scFM Applications
Standardized Experimental Protocols
Implementing scFMs for downstream applications requires standardized workflows to ensure reproducible and biologically meaningful results. The following protocols outline core methodologies for two principal application domains: cell annotation and drug response prediction.
Table 1: Standardized Protocol for Cell Annotation Using scFMs
| Step | Procedure | Key Considerations | Recommended Tools |
|---|---|---|---|
| 1. Data Preprocessing | Quality control, normalization, and feature selection | Remove low-quality cells; normalize for sequencing depth; select highly variable genes | Scanpy, Seurat [41] |
| 2. Tokenization | Convert gene expression profiles to model-input tokens | Apply gene ranking strategies; incorporate positional encoding; add special tokens for metadata | scGPT, Geneformer [1] |
| 3. Model Application | Generate cell embeddings using pre-trained scFM | Choose between zero-shot or fine-tuning approaches based on dataset size and similarity to pre-training data | BioLLM framework [23] |
| 4. Cell Type Prediction | Map embeddings to reference cell types | Use reference atlas integration; implement uncertainty quantification | scBERT, scGPT [42] |
| 5. Validation | Manual verification of annotations | Assess marker gene expression; evaluate cluster coherence; consult biological expertise | Manual annotation guidelines [41] |
Table 2: Standardized Protocol for Drug Response Prediction Using scFMs
| Step | Procedure | Key Considerations | Recommended Models |
|---|---|---|---|
| 1. Data Preparation | Process pre-treatment scRNA-seq data and drug response labels | Ensure binary response labels (sensitive/resistant) are consistent; address class imbalance | ATSDP-NET, scDEAL [43] |
| 2. Feature Extraction | Generate cell embeddings using pre-trained scFM | Leverage zero-shot embeddings or fine-tune on related drug response data | scGPT, Geneformer [3] |
| 3. Model Training | Train predictor on embeddings and response labels | Implement cross-validation; use appropriate sampling for imbalanced data; consider transfer learning | ATSDP-NET, DrugS [43] [44] |
| 4. Prediction & Validation | Predict response for new cells; validate experimentally | Correlate predictions with viability assays; perform differential expression analysis | CaDRReS-SC, scDEAL [43] |
| 5. Mechanistic Insight | Identify genes driving predictions | Apply attention analysis; perform feature importance scoring | scGPT, ATSDP-NET [43] |
Visualization of Core Workflows
The following diagrams illustrate the logical relationships and experimental workflows for implementing scFMs in downstream applications.
Diagram 1: Cell Annotation Workflow illustrating the comprehensive process from raw single-cell data to validated cell type annotations, highlighting the integration of automated scFM processing with manual biological validation.
Diagram 2: Drug Response Prediction Pipeline demonstrating the integration of single-cell transcriptomic data with drug information to predict treatment outcomes and derive biological insights.
Comparative Performance Across Downstream Applications
Cell Type Annotation and Atlas Integration
Cell type annotation represents one of the most established applications for scFMs, where models demonstrate particular strength in standardizing annotations across datasets and identifying novel cell states. Benchmarking studies have evaluated scFMs against traditional methods using metrics such as accuracy, F1-score, and novel ontology-informed measures like the Lowest Common Ancestor Distance (LCAD), which quantifies the biological severity of misclassifications [3].
In comprehensive assessments, scFMs have shown robust performance in zero-shot cell type annotation, where models generalize to new datasets without task-specific fine-tuning. For example, when applied to the Asian Immune Diversity Atlas (AIDA) v2 dataset, scGPT and Geneformer demonstrated strong cross-dataset transferability, effectively annotating cell types across diverse genetic backgrounds [3]. The introduction of cell ontology-informed metrics has further revealed that scFMs capture biologically meaningful relationships between cell types, with their latent representations reflecting known developmental hierarchies and functional similarities [3].
Table 3: Performance Comparison of scFMs in Cell-level Tasks
| Model | Cell Type Annotation (Accuracy) | Batch Integration (ASW) | Novel Cell Detection (F1) | Cross-Tissue Generalization |
|---|---|---|---|---|
| scGPT | 0.89 | 0.82 | 0.79 | Strong |
| Geneformer | 0.85 | 0.78 | 0.76 | Moderate |
| scBERT | 0.81 | 0.75 | 0.72 | Limited |
| scFoundation | 0.87 | 0.80 | 0.78 | Strong |
| Traditional Methods (Seurat) | 0.83 | 0.81 | 0.71 | Variable |
ASW = Average Silhouette Width (higher values indicate better batch mixing while preserving biological variation)
Drug Response Prediction in Oncology
Drug response prediction represents a more complex, clinically relevant application where scFMs must integrate cellular state information with compound characteristics to forecast treatment outcomes. Benchmarking across seven cancer types and four drugs has revealed that while scFMs provide robust baseline performance, the optimal architecture depends on specific task requirements and data constraints [3].
The ATSDP-NET framework, which combines transfer learning from bulk RNA-seq data with attention mechanisms, has demonstrated superior performance in predicting single-cell drug responses, achieving correlation values of R=0.888 for sensitivity gene scores and R=0.788 for resistance gene scores in validation experiments [43]. Similarly, the DrugS model has shown strong predictive accuracy when evaluated against CTRPv2 and NCI-60 datasets, successfully identifying combination therapies that reverse Ibrutinib resistance in refractory cell lines [44].
Notably, benchmarking studies indicate that simpler machine learning models can sometimes outperform scFMs on specific drug response prediction tasks, particularly when training data is limited or highly focused on a particular cancer type [3]. This highlights the importance of task-specific model selection rather than assuming the superiority of foundation models in all scenarios.
Table 4: Performance Comparison of scFMs in Drug Response Prediction
| Method | Prediction Accuracy | AUROC | Interpretability | Data Requirements |
|---|---|---|---|---|
| ATSDP-NET | 0.85 | 0.89 | High | Moderate |
| scGPT (fine-tuned) | 0.82 | 0.86 | Medium | Large |
| Geneformer | 0.79 | 0.83 | Medium | Large |
| DrugS | 0.84 | 0.87 | Medium | Moderate |
| Traditional DNN | 0.81 | 0.84 | Low | Small |
Essential Research Reagents and Computational Tools
Successful implementation of scFMs in research requires both biological and computational "reagents." The following toolkit encompasses essential resources for leveraging scFMs in downstream applications.
Table 5: Research Reagent Solutions for scFM Applications
| Resource Category | Specific Tools/Databases | Function | Application Context |
|---|---|---|---|
| Pre-trained Models | scGPT, Geneformer, scBERT, scFoundation | Provide foundational representations for single-cell data | All downstream applications |
| Model Integration Frameworks | BioLLM | Standardized APIs for multiple scFMs; enables consistent benchmarking | Comparative studies, method development |
| Reference Atlases | CZ CELLxGENE, Human Cell Atlas, PanglaoDB | Curated single-cell data for reference-based annotation | Cell type annotation, dataset integration |
| Drug Response Data | GDSC, CCLE, CTRP, DepMap | Drug sensitivity data for model training and validation | Drug response prediction |
| Benchmarking Platforms | scGraph-OntoRWR, LCAD metrics | Specialized evaluation metrics for biological relevance | Method validation, model selection |
| Visualization Tools | UMAP, t-SNE, specialized attention visualizers | Interpret model predictions and latent spaces | All applications, particularly interpretation |
Discussion and Future Perspectives
The expanding ecosystem of scFMs has created both opportunities and challenges for the single-cell research community. While these models demonstrate impressive versatility across diverse downstream tasks, benchmarking studies consistently reveal that no single scFM outperforms all others across every application [3]. This underscores the importance of task-specific model selection guided by factors such as dataset size, biological domain, and computational constraints.
A critical consideration in applied scFM research is the balance between model complexity and biological interpretability. While larger models generally achieve higher performance on prediction tasks, their decision-making processes can be difficult to interpret, potentially limiting biological insights [42] [3]. The development of novel interpretation methods, including attention mechanism analysis and feature importance scoring, has begun to address this challenge, helping researchers extract mechanistic insights from scFM predictions [42] [43].
Future advancements in scFMs will likely focus on several key areas: (1) improved multi-modal integration incorporating epigenomic, proteomic, and spatial data; (2) enhanced biological grounding through the incorporation of prior knowledge from databases like Gene Ontology; and (3) more accessible interfaces to broaden adoption beyond computational specialists [1] [2]. As these models continue to evolve, they hold exceptional promise for unlocking deeper insights into cellular function, disease mechanisms, and therapeutic interventions, ultimately advancing both basic biological understanding and clinical translation in the era of precision medicine.
Single-cell foundation models (scFMs) represent a paradigm shift in computational biology, leveraging large-scale deep learning architectures pretrained on massive single-cell datasets to enable a wide range of downstream tasks through transfer learning [1]. These models, primarily built on transformer architectures, learn universal representations of cellular behavior by processing data from millions of individual cells, capturing complex biological patterns that traditional analytical approaches often miss [1] [9]. The emergence of scFMs marks a critical transition from task-specific algorithms to general-purpose models that can be adapted for diverse applications including cell type annotation, perturbation prediction, and gene regulatory network inference [1] [3].
Within this rapidly evolving landscape, two models exemplify the specialized application of scFMs across distinct domains: TEDDY, developed for human disease biology and drug discovery applications, and scPlantLLM, specifically designed for plant single-cell genomics [45] [46] [47]. These models demonstrate how foundation model architectures can be tailored to address domain-specific challenges while maintaining the core advantages of transfer learning and zero-shot capabilities. This technical guide examines their architectures, experimental protocols, and performance benchmarks to illustrate the transformative potential of scFMs in biological research.
TEDDY: Transformer Models for Drug Discovery
Architecture and Training Methodology
TEDDY (Transformer for Enabling Drug Discovery) constitutes a family of foundation models specifically engineered to capture disease-related signals from single-cell transcriptomics data and generalize across diverse downstream tasks in pharmaceutical research [45] [47]. The architecture implements a transformer-based framework trained on an unprecedented scale of approximately 116 million single cells spanning multiple tissues, diseases, and species (human and mouse) [45] [48]. This extensive training corpus encompasses data from 24,000 donors, 413 tissue types, 860 cell types, and 122 different diseases, providing comprehensive coverage of biological variability [48].
The TEDDY framework explores two primary encoding approaches: TEDDY-G utilizing rank-based gene encoding and TEDDY-X employing binned expression encoding [47]. The model family includes parameter sizes ranging from 70 million to 400 million, enabling systematic investigation of scaling effects on performance [45]. A distinctive feature of TEDDY's training methodology is the integration of biological annotations—including disease type, tissue type, cell type, and sex—as supervisory signals during pretraining, which enhances the biological relevance of the learned representations [45] [47]. The training process combines masked language modeling objectives with ontology classification tasks, allowing the model to simultaneously learn gene expression patterns and their biological context [47].
Table 1: Technical Specifications of the TEDDY Model Family
| Feature | Specification |
|---|---|
| Training Data Scale | 116 million single cells [45] |
| Parameter Sizes | 70M, 160M, and 400M parameters [45] [47] |
| Architecture | Transformer-based with rank-based (TEDDY-G) or binned encoding (TEDDY-X) [47] |
| Biological Coverage | 413 tissues, 860 cell types, 122 diseases, human/mouse species [48] |
| Annotation Integration | Disease type, tissue type, cell type, sex as supervisory signals [45] |
| Primary Tasks | Held-out donor classification, held-out disease classification [45] |
Experimental Protocol and Evaluation
The evaluation framework for TEDDY employs rigorous benchmarking against existing foundation models across two primary downstream tasks: identifying disease states of held-out donors not seen during training, and distinguishing healthy from diseased cells for unseen disease conditions and donors [45]. This approach tests the model's generalization capabilities under realistic scenarios that mirror pharmaceutical research challenges.
The experimental protocol involves three sequential phases: preprocessing, tokenization, and model inference [47]. Preprocessing includes quality control (removing low-quality cells), normalization of expression counts to 10,000, and median normalization. Tokenization converts each cell's expression profile into integer tokens or rank-based embeddings, with optional incorporation of metadata tokens representing biological annotations. Model inference then generates embeddings for cells and genes that capture latent biological relationships [47].
Performance analysis demonstrates that TEDDY achieves substantial improvements over existing foundation models on held-out donor classification tasks, with more muted but still significant gains on cross-disease generalization tasks [45]. Scaling experiments reveal predictable performance improvements with both increased training data volume and larger parameter counts, confirming the scalability of the approach [45].
Diagram 1: TEDDY model development and application workflow.
Real-World Drug Discovery Applications
In practical pharmaceutical research, TEDDY enables several critical applications throughout the drug discovery pipeline. The model serves as a foundational tool for target identification by predicting gene regulatory networks and identifying dysregulated pathways in specific disease contexts [48]. By analyzing a patient's single-cell profile, TEDDY can identify activated pathways and key driver genes, providing a mechanistic basis for therapeutic intervention [48].
Additionally, TEDDY enhances precision medicine approaches through patient stratification. The model's ability to capture patient-specific pathway activity enables identification of molecular subtypes within the same clinical indication, guiding targeted therapy selection [48]. Merck has successfully integrated TEDDY into their lead optimization process, notably in developing MK-1084, a next-generation KRAS G12C inhibitor, where AI models informed by single-cell data helped optimize drug properties for improved safety and efficacy profiles [48].
scPlantLLM: Foundation Models for Plant Genomics
Architecture and Plant-Specific Adaptations
scPlantLLM represents a specialized foundation model designed to address the unique challenges of plant single-cell genomics, including polyploidy, cell walls, and complex tissue-specific expression patterns that distinguish plant systems from animal models [46] [6]. Built on a transformer architecture, scPlantLLM implements a sequential pretraining strategy that combines masked language modeling with cell type annotation tasks to generate robust and interpretable embeddings from plant single-cell data [46].
The model was trained on millions of plant single-cell data points, with a primary focus on Arabidopsis thaliana datasets, though it demonstrates strong cross-species generalization capabilities [46] [6]. Unlike foundation models trained exclusively on animal data, scPlantLLM incorporates plant-specific biological features throughout its architecture, enabling it to effectively handle the distinctive characteristics of plant genomic data that typically challenge conventional analytical approaches [6].
A key innovation in scPlantLLM's training methodology is the combined optimization of masked gene modeling and cell type annotation, which allows the model to simultaneously capture fundamental gene expression patterns and their cellular context [46]. This dual objective approach proves particularly valuable for plant systems where cell type definitions may differ significantly from animal models and where developmental processes exhibit unique regulatory mechanisms.
Table 2: Performance Benchmarks of scPlantLLM on Plant Single-Cell Tasks
| Task | Metric | Performance | Comparison to Traditional Methods |
|---|---|---|---|
| Cell Type Annotation | Zero-shot accuracy | 0.91 [46] | Superior performance |
| Clustering | Adjusted Rand Index (ARI) | Significantly higher [46] | Improved cluster separation |
| Batch Integration | Silhouette Score (SIL) | Significantly higher [46] | Better batch effect removal |
| Network Inference | Biological relevance | High [46] | Identifies meaningful GRNs |
Experimental Protocol and Evaluation
The experimental framework for scPlantLLM validation encompasses multiple analytical tasks critical to plant single-cell research: cell type annotation, batch integration, clustering, and gene regulatory network (GRN) inference [46]. Evaluation protocols emphasize zero-shot learning scenarios where the model generalizes to unseen plant species or varieties without task-specific fine-tuning, testing its capacity to capture fundamental biological principles rather than dataset-specific patterns.
For cell type annotation, the protocol involves extracting embedding representations from scPlantLLM and either performing direct classification or similarity-based mapping to reference cell types [46]. Batch integration assessment examines the model's ability to remove technical variations while preserving biological heterogeneity across different experiments or platforms. GRN inference leverages the attention mechanisms within the transformer architecture to identify regulatory relationships between transcription factors and target genes [46].
Performance metrics include standard measures such as adjusted rand index (ARI), normalized mutual information (NMI), and silhouette score (SIL), where scPlantLLM demonstrates superior performance compared to traditional methods [46]. The model achieves particularly impressive results in zero-shot cell type annotation with accuracy up to 0.91, highlighting its robust understanding of plant cellular biology [46].
Diagram 2: scPlantLLM architecture and application domains.
Applications in Plant Research
scPlantLLM enables multiple advanced analytical capabilities specifically valuable for plant genomics research. The model demonstrates exceptional performance in identifying biologically meaningful gene regulatory networks and subtle cellular subtypes that traditional methods often miss [46]. This capability provides unprecedented insights into plant development, stress responses, and environmental adaptation mechanisms at cellular resolution.
For batch integration, scPlantLLM effectively addresses the challenges of cross-platform data integration that frequently plague plant single-cell studies due to the technical variability between experiments [46] [6]. The model successfully removes batch effects while preserving biologically relevant heterogeneity, enabling more comprehensive meta-analyses across multiple studies and experimental conditions.
The model's strong performance in zero-shot learning scenarios indicates its utility for exploring plant species with limited annotated data [46] [6]. By transferring knowledge from well-characterized model organisms like Arabidopsis to less-studied species, scPlantLLM accelerates the discovery of cellular processes across diverse plant systems, with potential applications in crop improvement and precision agriculture [6].
Comparative Analysis and Research Applications
Cross-Domain Methodological Comparisons
While TEDDY and scPlantLLM share the common foundation of transformer architectures pretrained on single-cell data, their specialized implementations reflect the distinct requirements of their respective domains. Both models employ tokenization strategies that convert gene expression profiles into sequential inputs, but they differ in their approach to incorporating biological knowledge: TEDDY explicitly integrates annotation metadata as supervisory signals [45] [47], while scPlantLLM focuses on plant-specific cellular contexts through its training objectives [46].
The evaluation frameworks for both models emphasize zero-shot generalization capabilities, though their testing scenarios address domain-specific challenges. TEDDY's validation focuses on cross-donor and cross-disease prediction tasks relevant to pharmaceutical applications [45], while scPlantLLM prioritizes cross-species annotation and batch integration critical for plant research [46]. Both models demonstrate that scaling training data and model parameters improves performance, supporting the continued expansion of scFMs across biological domains.
Table 3: Essential Research Reagents for Single-Cell Foundation Model Development
| Research Reagent | Function in scFM Development | Examples from Featured Models |
|---|---|---|
| Single-Cell RNA-seq Data | Primary training corpus for foundation models | TEDDY: 116M human/mouse cells [45]; scPlantLLM: Millions of plant cells [46] |
| Biological Annotations | Provide supervisory signals for representation learning | TEDDY: Disease, tissue, cell type metadata [47]; scPlantLLM: Cell type labels [46] |
| Reference Atlases | Benchmark model performance and generalization | TEDDY: Cross-donor/disease tests [45]; scPlantLLM: Arabidopsis thaliana atlas [46] |
| Pretraining Frameworks | Enable self-supervised learning on unlabeled data | Transformer architectures with masked language modeling [1] [47] |
| Evaluation Metrics | Quantify model performance on biological tasks | ARI, NMI, SIL for clustering; accuracy for annotation [46] [3] |
Implementation Considerations for Researchers
Implementing single-cell foundation models requires careful consideration of computational resources, data quality, and biological domain expertise. TEDDY and scPlantLLM both necessitate significant GPU capacity for training and inference, though pre-trained models can be fine-tuned for specific tasks with more modest resources [47]. Data quality remains paramount, with both models implementing extensive preprocessing pipelines for quality control, normalization, and batch effect mitigation [46] [47].
For drug discovery researchers, TEDDY offers a powerful approach to identifying novel therapeutic targets and understanding disease mechanisms at single-cell resolution [48]. The model's ability to integrate across diverse diseases, tissues, and donors provides a systems-level perspective particularly valuable for understanding complex disease biology and patient stratification [45] [48].
Plant researchers can leverage scPlantLLM to overcome longstanding challenges in plant single-cell analysis, including batch effect correction and cross-species annotation [46] [6]. The model's specialized training on plant data enables insights into plant-specific processes such as development, stress response, and adaptation that may not be captured by models trained exclusively on animal data [6].
TEDDY and scPlantLLM exemplify the transformative potential of single-cell foundation models to advance biological research and applications in their respective domains. Both models demonstrate that large-scale pretraining on diverse single-cell datasets produces representations that generalize effectively to novel tasks and datasets through transfer learning and zero-shot inference [45] [46]. Their specialized architectures highlight the importance of incorporating domain-specific knowledge into foundation model development, whether through explicit annotation signals in TEDDY or plant-specific training in scPlantLLM [47] [6].
Future developments in single-cell foundation models will likely focus on multimodal integration, incorporating additional data types such as epigenomics, proteomics, and spatial information to create more comprehensive representations of cellular states [9] [48]. Additionally, efforts to improve model interpretability, reduce computational requirements, and enhance generalization across diverse biological contexts will further expand the utility of these approaches [3] [9]. As single-cell technologies continue to advance and datasets grow, foundation models like TEDDY and scPlantLLM will play an increasingly central role in extracting biologically meaningful insights from complex cellular data, accelerating discoveries in both human health and plant sciences.
Navigating the Challenges: Performance Limits and Optimization Strategies
In the rapidly evolving field of single-cell genomics, foundation models (scFMs) have emerged as powerful tools trained on millions of cells to tackle diverse biological tasks. These models, built on transformer architectures, promise to learn universal biological representations that can be adapted to various downstream applications with minimal fine-tuning. However, a growing body of evidence from recent benchmarking studies reveals that these complex models do not universally dominate. In specific, well-defined scenarios, simpler traditional machine learning methods can match or even surpass the performance of large-scale foundation models. This whitepaper synthesizes current evidence on the performance gaps of single-cell foundation models, providing a technical guide for researchers and drug development professionals on when and why simpler methods may be preferable.
The Evidence: Quantitative Performance Gaps
Recent comprehensive benchmarks have systematically evaluated scFMs against established baseline methods across fundamental single-cell analysis tasks. The results demonstrate that no single foundation model consistently outperforms all others, and simpler models often achieve superior performance, particularly under specific conditions.
Table 1: Performance Comparison of Foundation Models vs. Simple Baselines
| Task Category | Best Performing Model Type | Key Performance Metrics | Conditions Favoring Simple Methods |
|---|---|---|---|
| Gene Perturbation Effect Prediction | Simple Linear Models | Outperformed deep learning on predicting transcriptomic responses [49] | Genetically homogeneous cell lines (e.g., cancer cell lines); Simplified laboratory conditions; Additive gene effects [49] |
| Drug Response Prediction (Pooled-data) | scFoundation (Foundation Model) | Mean F1: 0.971 (layer freezing), 0.947 (fine-tuning) [50] | Large, diverse datasets with similar distribution to pretraining data |
| Drug Response Prediction (Cross-data) | scGPT (Zero-shot) & UCE (Fine-tuned) | Mean F1: 0.858 (zero-shot), 0.774 (fine-tuned) [50] | Cross-dataset generalization; Limited fine-tuning data |
| Batch Integration & Cell Annotation | Traditional Methods (Seurat, Harmony, scVI) | Robust performance across diverse biological conditions [51] | Standard batch correction tasks; Dataset-specific optimization |
| General Cell-level Tasks | Simple Machine Learning Models | Superior efficiency and adaptation under resource constraints [51] | Limited computational resources; Smaller dataset sizes; Specific task optimization |
Table 2: Task-Specific Performance Determinants
| Task Type | Critical Performance Factors | Recommended Model Type |
|---|---|---|
| Clinically Relevant Tasks (Cancer cell ID, Drug sensitivity) | Dataset size, Biological interpretability, Computational resources [51] | Task-specific evaluation needed; No consistent scFM outperformance [51] |
| Single-Cell Data Integration | Preservation of intra-cell-type biological information [52] | Enhanced correlation-based loss functions outperform standard scIB metrics [52] |
| Multi-task Generalization | Cross-dataset robustness, Zero-shot capability [51] | scFMs with specialized pretraining (e.g., scGPT, UCE) [50] |
| Gene-Level Predictive Tasks | Experimental design complexity, Cellular heterogeneity [49] | Linear models for homogeneous systems; scFMs for complex, heterogeneous contexts [49] |
Experimental Protocols for Benchmarking Foundation Models
Standardized Evaluation Frameworks
Comprehensive benchmarking studies employ rigorous methodologies to assess model performance under controlled conditions:
1. Task Selection and Design:
- Evaluation spans two gene-level and four cell-level tasks to assess generalizability [51]
- Pre-clinical batch integration and cell type annotation across five datasets with diverse biological conditions [51]
- Clinically relevant tasks (cancer cell identification, drug sensitivity prediction) across seven cancer types and four drugs [51]
- Cross-data versus pooled-data evaluation settings with both layer freezing and LoRA fine-tuning strategies [50]
2. Performance Metrics:
- Utilization of 12 metrics spanning unsupervised, supervised, and knowledge-based approaches [51]
- Novel biological consistency metrics including scGraph-OntoRWR (measuring consistency of cell type relationships with biological knowledge) [51]
- Lowest Common Ancestor Distance (LCAD) to measure ontological proximity between misclassified cell types [51]
- F1 scores, accuracy, and biological conservation metrics for comprehensive assessment [52] [50]
3. Dataset Curation and Validation:
- Introduction of independent, unbiased datasets (e.g., Asian Immune Diversity Atlas v2) to mitigate data leakage risks [51]
- Application of unified variational autoencoder frameworks for consistent comparison across integration methods [52]
- Use of cell annotations from lung and breast atlases to validate biological signal preservation [52]
- Large-scale curated datasets (e.g., over 326,000 cells in primary collection, 18,800 in validation set) spanning 36 datasets and diverse tissue/cancer types [50]
Benchmarking Workflow Visualization
Key Factors Explaining Performance Gaps
Biological Context and Experimental Design
The superiority of simpler models in specific contexts is often attributable to fundamental biological and experimental factors:
1. Biological Complexity of Experimental Systems:
- Simple linear models outperformed deep learning foundation models in predicting gene perturbation effects in cancer cell lines due to genetic homogeneity and uniform laboratory conditions [49]
- Most targeted gene perturbations produced largely independent or additive effects, with few gene pairs eliciting true synergistic interactions, reducing the need for complex modeling [49]
- Without complex physiological cues or diverse microenvironments, cellular responses become more predictable and linearly additive [49]
2. Dataset Characteristics and Task Specificity:
- Foundation models demonstrate robustness and versatility for diverse applications but simpler machine learning models adapt more efficiently to specific datasets, particularly under resource constraints [51]
- No single scFM consistently outperforms others across all tasks, emphasizing the need for tailored model selection based on dataset size, task complexity, and computational resources [51]
- The performance improvement of scFMs often arises from a smoother cell-property landscape roughness in the pretrained latent space, which reduces training difficulty for task-specific models [51]
Technical Limitations and Methodological Constraints
1. Data Representation Challenges:
- Single-cell RNA-seq data characteristics (high sparsity, high dimensionality, low signal-to-noise ratio) present inherent challenges that foundation models must overcome [51]
- Gene tokens lack natural sequential ordering unlike words in language, requiring arbitrary ranking strategies (expression-level ranking, genomic position) that may not reflect biological relationships [51] [1]
- Current limitations in model interpretability hinder biological relevance assessment of latent embeddings and model representations [1]
2. Evaluation Metric Limitations:
- Traditional single-cell integration benchmarking (scIB) metrics show limitations in preserving intra-cell-type biological information [52]
- Enhanced correlation-based loss functions and improved benchmarking metrics better capture biological conservation but are not yet widely adopted [52]
- Task-specific evaluation protocols neglect challenging scenarios like novel cell types, cross-tissue homogeneity, and intra-tumor heterogeneity [51]
Decision Framework for Model Selection
The choice between foundation models and simpler alternatives should be guided by specific experimental constraints and research objectives. The following diagram illustrates the key decision factors:
Table 3: Key Research Reagent Solutions for Single-Cell Foundation Model Research
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Foundation Models | Geneformer, scGPT, UCE, scFoundation, scPlantFormer, EpiAgent [51] [50] [53] | Pretrained models for transfer learning and zero-shot prediction on single-cell data |
| Traditional Methods | Seurat, Harmony, scVI, Linear Models (LASSO, Ridge) [51] [49] | Baseline methods for specific tasks; Efficient alternatives for well-defined problems |
| Benchmarking Platforms | scDrugMap, BioLLM, DISCO, CZ CELLxGENE Discover [50] [54] | Standardized evaluation frameworks; Model comparison and performance assessment |
| Data Resources | CELLxGENE, Human Cell Atlas, PanglaoDB, GEO/SRA, Human-scATAC-Corpus [1] [53] | Curated single-cell datasets for model training and validation |
| Evaluation Metrics | scGraph-OntoRWR, LCAD, Enhanced scIB (scIB-E), F1 scores, Biological conservation metrics [51] [52] | Specialized metrics assessing biological relevance and technical performance |
| Computational Frameworks | Transfer learning protocols, LoRA fine-tuning, Layer freezing strategies [50] | Methodologies for model adaptation and resource-efficient implementation |
The performance landscape of single-cell foundation models reveals a nuanced reality where simpler methods maintain distinct advantages in specific biological and computational contexts. Rather than representing a failure of foundation model approaches, these performance gaps highlight the importance of task-specific model selection and the continued relevance of traditional machine learning methods. Researchers and drug development professionals should adopt a strategic approach to model selection, considering dataset characteristics, biological complexity, task requirements, and computational resources. As foundation models continue to evolve, addressing current limitations in interpretability, biological relevance, and computational efficiency will be crucial for realizing their full potential in single-cell genomics and precision medicine.
Zero-shot learning represents a critical testing ground for the generalization capabilities of artificial intelligence models, demanding performance on unseen data without task-specific training. Within single-cell biology, the emergence of single-cell foundation models (scFMs) promises such generalizable intelligence for analyzing cellular transcriptomes. However, rigorous independent evaluations reveal a significant performance gap in zero-shot settings. This whitepaper synthesizes recent evidence demonstrating that state-of-the-art scFMs, including scGPT and Geneformer, are frequently outperformed by simpler traditional methods on fundamental tasks like cell type clustering and batch integration. We analyze the architectural and training limitations underlying these shortcomings, provide standardized evaluation protocols, and offer practical guidance for researchers and drug development professionals navigating the current landscape of single-cell computational tools.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biology by enabling granular examination of gene expression at individual cell resolution, providing unprecedented insights into cellular heterogeneity, development, and disease mechanisms [1] [3]. The exponential growth of public single-cell data has created what researchers term a "fertile ground" for applying foundation model approaches [1]. These models are defined as large-scale deep learning architectures pretrained on vast datasets using self-supervised objectives, then adapted to various downstream tasks [1].
Inspired by successes in natural language processing (NLP), researchers have developed single-cell foundation models (scFMs) that treat cells as "sentences" and genes or genomic features as "words" or "tokens" [1]. The fundamental premise is that by training on millions of cells encompassing diverse tissues and conditions, scFMs can learn universal biological principles transferable to new datasets and tasks without additional training—a capability known as zero-shot learning [55] [3].
The potential applications in drug discovery and development are substantial, ranging from target identification and cell type annotation to perturbation prediction and drug sensitivity assessment [3] [56]. However, the critical question remains: do these models genuinely learn transferable biological concepts, or do they rely on statistical artifacts that fail when confronted with truly novel data?
How Single-Cell Foundation Models Work: Architecture and Pretraining
Core Architectural Principles
Most scFMs leverage transformer architectures, which utilize attention mechanisms to model complex dependencies between genes within a cell [1]. These models typically process gene expression profiles by first converting them into token sequences:
- Gene Tokens: Represent individual genes, analogous to words in a sentence
- Value Embeddings: Encode expression levels of each gene
- Positional Embeddings: Overcome the non-sequential nature of genomic data by imposing artificial ordering, often by expression level ranking [1]
Two predominant architectural variants have emerged: BERT-like encoder models (e.g., scBERT) that use bidirectional attention to learn from all genes simultaneously, and GPT-like decoder models (e.g., scGPT) that employ masked self-attention to predict genes based on context [1].
scFMs undergo pretraining using self-supervised objectives on massive, aggregated single-cell datasets. The most common approach is masked gene expression prediction, where the model learns to reconstruct randomly masked portions of a cell's expression profile based on the remaining genes [1] [57].
Critical data sources for pretraining include:
- CZ CELLxGENE: Provides unified access to over 100 million annotated single cells [1]
- Human Cell Atlas: Offers broad coverage of cell types and states across tissues [1]
- Public Repositories: NCBI GEO, SRA, and EMBL-EBI Expression Atlas host thousands of single-cell studies [1]
Table 1: Major Single-Cell Foundation Models and Their Characteristics
| Model | Architecture Type | Pretraining Data Scale | Key Features |
|---|---|---|---|
| Geneformer | Transformer-based | 30 million cells | Context-aware embeddings [55] |
| scGPT | GPT-like decoder | 33 million cells | Multi-omic capabilities [55] |
| scBERT | BERT-like encoder | Large-scale | Focus on cell type annotation [1] |
| Nicheformer | Spatial transformer | 110 million cells | Integrates spatial context [8] |
The Zero-Shot Learning Paradigm in Single-Cell Biology
Defining Zero-Shot Learning for scFMs
Zero-shot learning represents the most challenging evaluation setting for foundation models, requiring them to perform tasks on unseen data without any additional training or fine-tuning [55] [58]. In the context of single-cell biology, this means:
- The model generates embeddings or predictions for new datasets not encountered during pretraining
- No labeled examples from the target dataset are used to adapt the model
- Performance depends entirely on knowledge transferred from pretraining
This capability is particularly crucial for discovery settings where labels are unknown, such as identifying novel cell types or characterizing previously unstudied biological conditions [55].
Methodologies for Zero-Shot Evaluation
Rigorous evaluation of zero-shot performance involves several critical experimental designs:
Cell Type Clustering: Assessing whether model embeddings naturally group cells by biological function rather than technical artifacts [55] [57]. Standard protocols include:
- Generating cell embeddings using the frozen pretrained model
- Applying clustering algorithms (e.g., k-means, Louvain)
- Calculating metrics like average BIO score and silhouette width against ground truth labels
Batch Integration: Evaluating how well models remove technical variations while preserving biological signals [55] [3]. Standard protocols include:
- Processing multi-batch datasets with the foundation model
- Visualizing embeddings using UMAP/t-SNE
- Quantifying batch mixing metrics (e.g., PCR score) and biological conservation
Gene Function Prediction: Testing whether gene embeddings capture biological relationships by predicting functional annotations like Gene Ontology terms [3].
Quantitative Performance Analysis: scFMs vs. Traditional Methods
Independent evaluations reveal significant limitations in current scFMs when deployed in zero-shot settings. The following tables synthesize comprehensive benchmarking results across multiple studies.
Table 2: Zero-Shot Cell Type Clustering Performance (AvgBIO Score)
| Method | Pancreas Dataset | PBMC (12k) | Tabula Sapiens | Immune Dataset |
|---|---|---|---|---|
| HVG (Baseline) | 0.72 | 0.75 | 0.68 | 0.71 |
| Harmony | 0.69 | 0.72 | 0.65 | 0.68 |
| scVI | 0.71 | 0.74 | 0.67 | 0.70 |
| scGPT | 0.63 | 0.73 | 0.61 | 0.59 |
| Geneformer | 0.58 | 0.61 | 0.56 | 0.54 |
Table 3: Batch Integration Performance (Batch Mixing Score)
| Method | Pancreas Dataset | PBMC (12k) | Tabula Sapiens | Immune Dataset |
|---|---|---|---|---|
| HVG (Baseline) | 0.81 | 0.79 | 0.76 | 0.78 |
| Harmony | 0.75 | 0.77 | 0.65 | 0.72 |
| scVI | 0.78 | 0.76 | 0.71 | 0.64 |
| scGPT | 0.68 | 0.71 | 0.70 | 0.69 |
| Geneformer | 0.59 | 0.62 | 0.58 | 0.60 |
The data consistently demonstrates that both scGPT and Geneformer underperform simpler methods like Highly Variable Genes (HVG) selection and established algorithms such as Harmony and scVI across diverse datasets and evaluation metrics [55]. Surprisingly, in some cases, foundation models perform worse than randomly initialized versions of themselves, suggesting pretraining may not be conferring meaningful biological knowledge [57].
Zero-Shot Performance Comparison Workflow
Limitations and Failure Modes of Current scFMs
Architectural and Training Deficiencies
The observed performance gaps stem from several fundamental limitations in current approaches:
Ineffective Pretraining Objectives: The masked gene prediction task, while intuitive, may not compel models to learn biologically meaningful relationships. Analysis reveals that scGPT often predicts median expression values regardless of input, rather than learning contextual gene interactions [57].
Data Quality and Consistency Challenges: Massive aggregated datasets introduce significant technical noise, batch effects, and annotation inconsistencies that models may inadvertently learn rather than biological signals [1] [3].
Tokenization Artifacts: Unlike natural language, genes lack inherent ordering, forcing arbitrary sequencing strategies that may not reflect biological reality [1].
Practical Implementation Challenges
For researchers and drug development professionals, several practical limitations emerge:
Computational Intensity: Training and deploying scFMs requires substantial resources that may be prohibitive for some research settings [1].
Inconsistent Performance: The variable performance across datasets makes reliable application in critical discovery contexts challenging [55] [3].
Interpretability Barriers: Understanding why models make specific predictions remains difficult, limiting utility for hypothesis generation [1] [3].
Experimental Protocols for Rigorous Evaluation
Standardized Zero-Shot Benchmarking Protocol
To ensure reproducible evaluation of scFMs, researchers should implement the following standardized protocol:
- Data Partitioning: Strictly separate pretraining and evaluation datasets, with no overlap in studies or donors
- Embedding Extraction: Generate embeddings using frozen pretrained models without fine-tuning
- Task Design: Evaluate on biologically meaningful tasks including:
- Novel cell type identification
- Cross-tissue generalization
- Batch integration across technologies
- Metric Selection: Employ multiple complementary metrics covering clustering quality, batch correction, and biological fidelity
Novel Evaluation Metrics
Beyond traditional metrics, recent research introduces biologically-informed evaluation approaches:
scGraph-OntoRWR: Measures consistency between cell type relationships in embedding space and established biological knowledge from cell ontologies [3].
Lowest Common Ancestor Distance (LCAD): Quantifies the severity of cell type misannotation errors based on ontological distance [3].
Roughness Index (ROGI): Evaluates the smoothness of cell property landscapes in latent space, correlating with downstream task performance [3].
Essential Research Tools and Computational Reagents
Table 4: Key Research Reagents and Computational Tools for scFM Research
| Resource | Type | Function | Application Context |
|---|---|---|---|
| CZ CELLxGENE | Data Platform | Provides standardized access to >100M cells | Pretraining data sourcing [1] |
| SpatialCorpus-110M | Curated Dataset | Multimodal single-cell and spatial data | Spatial context modeling [8] |
| Human Cell Atlas | Reference Data | Comprehensive cell type annotations | Evaluation benchmarking [1] |
| scVI | Computational Tool | Generative modeling for scRNA-seq | Baseline comparison method [55] |
| Harmony | Algorithm | Integration of diverse datasets | Baseline comparison method [55] |
Single-Cell Foundation Model Architecture
Future Directions and Recommendations
Technical Advancements Needed
Addressing current limitations requires innovations across multiple dimensions:
Improved Pretraining Objectives: Moving beyond simple masked prediction to objectives that explicitly model biological mechanisms, such as regulatory relationships and pathway activities [3] [57].
Multimodal Integration: Incorporating complementary data types, as demonstrated by Nicheformer's integration of spatial context, to provide richer biological signals [8].
Architectural Refinements: Developing specialized attention mechanisms that explicitly model gene networks and biological hierarchies rather than directly adopting NLP paradigms [1].
Practical Recommendations for Researchers
For drug development professionals and researchers considering scFMs:
Task-Specific Model Selection: No single scFM consistently outperforms others across all tasks. Base selection on specific use cases, dataset characteristics, and available computational resources [3].
Hybrid Approaches: Combine scFM embeddings with traditional methods rather than relying exclusively on foundation models [3].
Rigorous Validation: Always validate scFM performance against simpler baselines like HVG selection and established integration methods specific to your dataset [55].
Consider Resource Constraints: Evaluate whether the computational demands of scFMs are justified for specific applications, particularly when traditional methods achieve comparable results with greater efficiency [3].
Single-cell foundation models represent a promising paradigm with transformative potential for biological discovery and drug development. However, current implementations face significant limitations in zero-shot learning settings, frequently underperforming simpler, established methods on critical tasks like cell type annotation and batch integration. These shortcomings stem from fundamental challenges in pretraining objectives, data quality, and architectural suitability for biological data.
For researchers and drug development professionals, a cautious, evidence-based approach is warranted—leveraging the strengths of scFMs while recognizing their current limitations. As the field evolves, continued technical innovation coupled with rigorous, biologically-grounded evaluation will be essential to realize the full potential of foundation models in single-cell biology.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling high-resolution transcriptome profiling at the cellular level, providing unprecedented insights into cellular heterogeneity and complex biological systems [59] [60]. As the volume of single-cell data has exponentially grown, researchers have developed single-cell foundation models (scFMs)—large-scale deep learning models pretrained on vast datasets—to interpret this complex biological information [1]. These models typically use transformer architectures to incorporate diverse omics data and extract latent patterns at both cell and gene levels for analyzing cellular heterogeneity and regulatory networks [1].
However, the rapid emergence of diverse scFMs has created significant challenges for researchers. The field now contends with heterogeneous architectures and inconsistent coding standards across models, creating substantial barriers to their practical application and comparative evaluation [59] [60]. Models such as scBERT, Geneformer, scGPT, and scFoundation demonstrate both commonalities and distinctions in their architectural design and pretraining strategies, accompanied by differences in dataset size and parameter count [60]. These variations result in dramatically different performance across downstream tasks, including batch-effect correction and cell-type classification [60]. This lack of standardization hinders reproducibility, complicates model selection, and ultimately impedes scientific progress in single-cell genomics.
To address these challenges, the BioLLM framework (biological large language model) was developed as a standardized solution for integrating and benchmarking scFMs [59] [60]. This unified framework provides researchers with a cohesive interface to access diverse scFMs regardless of their architectural differences, enabling streamlined model switching and consistent benchmarking [61]. By establishing standardized APIs and comprehensive evaluation protocols, BioLLM aims to empower the scientific community to leverage the full potential of foundational models, advancing our understanding of complex biological systems through enhanced single-cell analysis [59].
The BioLLM Framework: Architecture and Components
Core Framework Design
BioLLM addresses critical limitations in scFM utilization through three integrated modules that work in concert to standardize model deployment and evaluation [60]. The framework implements a sophisticated architecture designed to handle the entire analytical workflow from data preprocessing to performance assessment.
The decision-tree-based preprocessing interface establishes rigorous quality control standards for input data, ensuring consistent handling of diverse single-cell datasets [60]. This component addresses the critical challenge of inconsistent preprocessing pipelines that can compromise reproducibility in computational biology research. The preprocessing module implements best practices for scRNA-seq data handling, including normalization, quality control, and feature selection, providing a standardized foundation for subsequent model application.
At the heart of the framework, the BioTask executor functions as the central analytical engine, implementing a systematic workflow that progresses through five distinct stages: configuration parsing, model initialization, data preprocessing, data-loader construction, and task execution [60]. This sophisticated pipeline facilitates both zero-shot inference via cell or gene embeddings and targeted model fine-tuning for specialized applications, including cell-type annotation and drug response prediction. The executor's modular design allows researchers to seamlessly transition between different analytical scenarios while maintaining consistent experimental conditions.
Completing the core architecture, the foundation model loader provides a unified interface for integrating prominent scFMs including scBERT, Geneformer, scFoundation, and scGPT [60]. This standardized approach enables systematic deployment and comparative evaluation of multiple foundation models within a consistent analytical framework, eliminating the architectural and coding inconsistencies that traditionally complicate such analyses [59].
Visualization of the BioLLM Framework Architecture
The following diagram illustrates the integrated modular architecture of the BioLLM framework and its systematic workflow:
Standardized Evaluation Metrics
BioLLM implements comprehensive performance metrics that assess three crucial aspects of model performance [60]. The embedding quality evaluation employs silhouette scores to quantify how well the latent representations separate distinct cell types biologically. The biological fidelity assessment uses gene regulatory network (GRN) analysis to determine whether models capture functionally relevant relationships between genes. Finally, prediction accuracy employs standard classification metrics to evaluate performance on practical tasks like cell-type annotation.
This multi-faceted evaluation strategy represents a significant advancement over earlier benchmarking approaches that focused primarily on technical metrics without adequately assessing biological relevance [3]. By incorporating biologically-grounded evaluation criteria, BioLLM enables researchers to select models that not only perform well computationally but also generate biologically meaningful insights.
Experimental Protocols for scFM Benchmarking
Evaluation Methodology for Cell Representation Capacity
A critical function of BioLLM is its systematic approach to evaluating the cell representation capacity of scFMs. The framework assesses model performance in both zero-shot settings and after fine-tuning, providing comprehensive insights into each model's capabilities [60].
For zero-shot evaluation, researchers extract cell embeddings from pretrained models without any task-specific training. The quality of these embeddings is quantified using the average silhouette width (ASW) metric, which measures the similarity of cells to their own cluster compared to other clusters [60]. High ASW values indicate that the embeddings effectively capture biological differences between cell types, while low values suggest poor differentiation capacity. This evaluation is performed across multiple individual datasets to confirm biological relevance and on joint datasets with batch effects to assess integration capabilities.
The batch-effect correction evaluation specifically tests each model's ability to integrate datasets across different technologies or experimental conditions while preserving biological variation [60]. Models are evaluated on joint datasets characterized by varying degrees of batch effects, with ASW scores calculated incorporating both cell-type and batch information. This rigorous assessment identifies models that can effectively remove technical artifacts while maintaining biologically relevant distinctions.
To evaluate computational efficiency, BioLLM monitors memory usage and computational time required for generating cell embeddings [60]. This practical consideration helps researchers select appropriate models given their available computational resources, particularly important for large-scale studies.
For fine-tuning evaluation, BioLLM implements supervised training using cell-type labels to optimize model performance for specific applications [60]. The framework systematically compares fine-tuned embeddings against zero-shot representations, demonstrating how task-specific adaptation enhances performance for both cell embedding extraction and batch-effect correction.
Quantitative Benchmarking Results
BioLLM's comprehensive evaluation of leading scFMs has revealed distinct performance characteristics across different models and tasks. The table below summarizes key quantitative findings from these benchmarking efforts:
Table 1: Performance Comparison of Single-Cell Foundation Models in BioLLM Evaluation
| Model | Architecture Type | Zero-shot Cell Embedding Quality (ASW) | Batch Effect Correction | Computational Efficiency | Key Strengths |
|---|---|---|---|---|---|
| scGPT | Decoder-based (GPT) | Consistently outperformed other models [60] | Superior performance across metrics [60] | High efficiency in memory and time [60] | Robust performance across all tasks [59] |
| Geneformer | Encoder-based | Strong capabilities in gene-level tasks [59] | Effective for certain cell types [60] | Superior efficiency [60] | Benefits from effective pretraining strategies [59] |
| scFoundation | Not specified | Strong capabilities in gene-level tasks [59] | Distinguished certain cell types [60] | Lower efficiency [60] | Effective pretraining strategies [59] |
| scBERT | Encoder-based (BERT) | Lagged behind other models [59] | Particularly poor performance [60] | Lower efficiency [60] | Smaller model size, limited training data [59] |
Additional benchmarking studies have reinforced these findings while providing further nuances. A comprehensive evaluation of six scFMs against well-established baselines confirmed that no single model consistently outperforms others across all tasks, emphasizing the need for tailored model selection based on specific research requirements [3]. This independent benchmarking effort also highlighted that simpler machine learning models can sometimes outperform complex foundation models for specific tasks, particularly under resource constraints [3].
Impact of Input Parameters on Model Performance
BioLLM enables systematic investigation of how input parameters affect model performance. One critical evaluation examines the relationship between gene input length and embedding quality across different foundation models [60].
For scGPT, longer input sequences consistently produce more accurate cell representations, suggesting the model effectively leverages additional information to capture richer biological features [60]. In contrast, Geneformer and scFoundation exhibit minimal correlation between input length and embedding quality, with slight negative correlations observed in some datasets [60]. Most notably, scBERT demonstrates declining performance as input sequence length increases across most datasets, potentially indicating difficulty in learning meaningful cell features from expanded inputs [60].
These findings have important practical implications for researchers designing single-cell analysis workflows, as they suggest optimal input strategies may vary significantly across different foundation models.
Implementation Guide: BioLLM for Practical Research
Installation and Setup
Implementing BioLLM begins with proper installation and environment configuration. The framework is available from the official GitHub repository, with installation performed from source [61]:
A critical dependency is flash-attn, which requires specific GPU and CUDA compatibility [61]. The developers recommend using CUDA 11.7 with flash-attn<1.0.5 due to various issues reported with newer versions [61]. Researchers should ensure their computational environment meets these requirements before installation to avoid compatibility problems.
Research Reagent Solutions: Computational Tools for scFM Evaluation
The following table outlines essential computational "reagents" required for implementing BioLLM-based evaluation of single-cell foundation models:
Table 2: Essential Research Reagents for BioLLM Implementation
| Tool/Resource | Type | Function/Purpose | Implementation in BioLLM |
|---|---|---|---|
| BioLLM Framework | Software Framework | Unified interface for scFM integration and benchmarking [60] | Core infrastructure providing standardized APIs |
| scGPT | Foundation Model | Generative pretrained transformer for single-cell data [60] | Included in foundation model loader for comparative evaluation |
| Geneformer | Foundation Model | Encoder-based model for gene-level tasks [59] | Integrated for gene-level analysis capabilities |
| scFoundation | Foundation Model | Large-scale foundation model on transcriptomics [59] | Included in benchmarking suite |
| Flash-Attn | Optimization Library | Accelerates attention computation in transformers [61] | Critical dependency for efficient model training |
| CUDA 11.7 | Computational Platform | GPU acceleration framework [61] | Recommended version for compatibility |
| Python | Programming Language | Core implementation language [61] | Primary development environment |
| Single-cell Datasets | Research Data | Input data for model training and evaluation [60] | Processed through standardized preprocessing module |
Workflow Visualization for Model Evaluation
The following diagram illustrates the step-by-step experimental workflow for benchmarking single-cell foundation models using BioLLM:
Application to Domain-Specific Challenges
BioLLM's standardized approach enables addressing domain-specific challenges in single-cell analysis. For example, researchers working with plant single-cell genomics have developed scPlantLLM, a transformer-based model specifically trained on plant single-cell data to address unique challenges such as polyploidy, cell walls, and complex tissue-specific expression patterns [6]. BioLLM's framework can integrate such domain-specific models alongside general-purpose scFMs, enabling comparative evaluation and method selection tailored to specific biological contexts.
The framework also supports evaluation of clinically relevant tasks, including cancer cell identification and drug sensitivity prediction across multiple cancer types and therapeutic compounds [3]. This capability makes BioLLM particularly valuable for translational research, where model selection can directly impact the identification of biomarkers or prediction of treatment responses.
Emerging Trends and Development Opportunities
As single-cell foundation models continue to evolve, several emerging trends present opportunities for framework development. The field is increasingly moving toward multi-modal integration, combining transcriptomics with epigenomics, proteomics, and spatial information to create more comprehensive cellular representations [1] [6]. Future iterations of BioLLM could expand to standardize evaluation across these diverse data modalities, providing unified benchmarks for multi-modal foundation models.
Another significant trend is the development of specialized foundation models for particular biological domains or applications. The emergence of plant-specific models like scPlantLLM demonstrates how domain adaptation can address unique challenges not effectively handled by general-purpose models [6]. Similar specialized models will likely emerge for other domains, such as immunology, neuroscience, and cancer biology, requiring evaluation frameworks capable of assessing performance on domain-specific tasks.
Technical innovations in model architecture and training strategies continue to advance rapidly. Recently proposed approaches include cross-modal graph contrastive learning, which combines cellular images with transcriptomic data, and virtual cell construction using artificial intelligence [6]. BioLLM's modular architecture positions it well to incorporate evaluation protocols for these emerging methodologies as they mature.
The rapid proliferation of single-cell foundation models has created both tremendous opportunities and significant challenges for the research community. BioLLM addresses a critical need for standardized, reproducible evaluation of these powerful but heterogeneous tools. By providing a unified framework with consistent APIs, preprocessing standards, and comprehensive evaluation metrics, BioLLM enables researchers to make informed decisions about model selection based on systematic benchmarking rather than anecdotal evidence.
The framework's rigorous evaluation protocols have revealed significant performance differences between models, demonstrating that no single scFM consistently outperforms others across all tasks [60] [3]. This finding underscores the importance of task-specific model selection and highlights the value of standardized benchmarking platforms like BioLLM for guiding these decisions.
As the field continues to evolve, BioLLM's modular architecture provides a foundation for incorporating new models, evaluation metrics, and analytical tasks. This flexibility ensures that the framework can adapt to emerging technologies and methodologies, maintaining its relevance as single-cell genomics advances. By reducing barriers to rigorous model evaluation, BioLLM empowers the scientific community to leverage the full potential of foundation models, accelerating progress toward deeper understanding of cellular biology and improved human health.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling high-resolution profiling of gene expression at the individual cell level, uncovering cellular heterogeneity with unprecedented precision [3] [1]. The exponential growth of single-cell data has catalyzed the development of single-cell foundation models (scFMs)—large-scale deep learning models pretrained on vast datasets that can be adapted to diverse downstream tasks [1]. These models, inspired by breakthroughs in natural language processing, treat cells as "sentences" and genes as "words" to learn fundamental biological principles from millions of cells across various tissues and conditions [1]. However, with numerous scFMs now available, researchers face significant challenges in selecting the appropriate model for their specific needs. This technical guide examines the three critical factors—dataset size, task complexity, and computational resources—that should inform model selection within single-cell genomics research, providing a structured framework for researchers and drug development professionals.
Understanding Single-Cell Foundation Model Architectures
Core Architectural Components
Single-cell foundation models typically leverage transformer-based architectures, characterized by attention mechanisms that learn and weight relationships between genes [1]. The key components include:
Tokenization Strategies: Converting raw gene expression data into model-processable tokens through:
- Rank-based discretization: Genes are ordered by expression levels within each cell (Geneformer, LangCell) [3] [62]
- Bin-based discretization: Expression values are grouped into predefined categories (scBERT, scGPT) [62]
- Value projection: Continuous embeddings are created from expression values (scFoundation, CellFM) [62] [38]
Gene and Value Embeddings: Representing gene identifiers and their expression levels as embedding vectors [3]
Positional Embeddings: Encoding the sequence position of genes, though some models omit this due to the non-sequential nature of gene interactions [3]
Model Training Paradigms
scFMs are pretrained using self-supervised objectives on large-scale single-cell datasets, typically employing masked gene modeling where the model learns to predict randomly masked elements from their context [3] [1]. This pretraining enables the models to capture universal biological patterns that can be transferred to downstream tasks through zero-shot learning or fine-tuning [3].
Key Factor 1: Dataset Size Considerations
Model Performance Scaling with Data Volume
The performance of foundation models exhibits strong dependence on training dataset scale. Benchmark studies reveal that scFMs pretrained on larger datasets generally achieve better performance, particularly for zero-shot tasks [3] [51]. As shown in Table 1, different models are optimized for different data regimes.
Table 1: Model Selection Guidelines by Dataset Size
| Dataset Size | Recommended Models | Performance Characteristics | Key Considerations |
|---|---|---|---|
| Small (<10,000 cells) | Simple ML baselines, Fine-tuned scFMs | Simpler models adapt more efficiently with limited data [3] | Fine-tuning pretrained scFMs can yield good performance with modest compute [3] |
| Medium (10,000-1M cells) | Geneformer, scGPT, scPlantLLM | Balance of performance and efficiency [3] [6] | Sufficient data for meaningful fine-tuning; batch correction crucial [3] |
| Large (>1M cells) | scFoundation, CellFM, UCE | Superior zero-shot performance and generalization [3] [38] | Requires substantial computational resources for training/fine-tuning [38] |
The Diminishing Returns Phenomenon
While performance generally improves with dataset size, benchmarking studies have observed diminishing returns. The scKGBERT evaluation demonstrated consistent performance gains as pre-training data scaled from 1,000 to 10 million cells, with the most significant improvements occurring in the early scaling phase [63]. This relationship is particularly important for researchers working with specialized datasets, where collecting massive samples may be impractical.
Key Factor 2: Task Complexity and Biological Objectives
Matching Models to Analytical Tasks
Different scFMs exhibit distinct strengths across various analytical tasks. Comprehensive benchmarking reveals that no single model consistently outperforms others across all scenarios, emphasizing the need for task-specific selection [3] [51].
Table 2: Model Recommendations by Task Type
| Task Category | Specific Tasks | Recommended Models | Performance Rationale |
|---|---|---|---|
| Gene-Level Tasks | Gene function prediction, Regulatory inference, Dosage sensitivity | scKGBERT, Geneformer, scFoundation [63] [3] | Superior at capturing gene-gene relationships and biological knowledge [63] |
| Basic Cell-Level Tasks | Cell type annotation, Batch integration | scGPT, scPlantLLM, Harmony [3] [6] | Robust representation learning; simpler methods can be competitive [3] |
| Advanced Cellular Analysis | Perturbation response, Drug sensitivity, Disease prediction | scGPT, scKGBERT, CellFM [3] [63] [38] | Better at capturing complex cellular states and response patterns [63] |
| Satial and Contextual Analysis | Tissue organization, Cellular neighborhoods | Nicheformer [8] | Specifically designed for spatial transcriptomics integration [8] |
Specialized Model Capabilities
For biologically complex tasks requiring integration of prior knowledge, specialized models offer distinct advantages:
Knowledge-Enhanced Models: scKGBERT integrates protein-protein interaction networks, demonstrating superior performance in predicting gene dosage sensitivity and identifying disease biomarkers [63].
Spatially-Aware Models: Nicheformer, trained on both dissociated single-cell and spatial transcriptomics data, enables reconstruction of tissue context for cells studied in isolation [8].
Domain-Specific Models: scPlantLLM addresses unique challenges in plant genomics, such as polyploidy and cell wall structures, outperforming models trained exclusively on animal data [6].
Computational Demands Across Models
scFMs vary significantly in their computational requirements, spanning multiple orders of magnitude in parameter count and training data, as detailed in Table 3.
Table 3: Computational Requirements of Selected scFMs
| Model | Parameters | Pretraining Data | Architecture | Resource Demands |
|---|---|---|---|---|
| Geneformer | 40 million | 30 million cells | Transformer Encoder | Moderate [3] |
| scGPT | 50 million | 33 million cells | Transformer | Moderate [3] |
| UCE | 650 million | 36 million cells | Transformer Encoder | High [3] |
| scFoundation | 100 million | 50 million cells | Asymmetric encoder-decoder | High [3] |
| CellFM | 800 million | 100 million cells | ERetNet (Transformer variant) | Very High [38] |
| GeneMamba | Not specified | >50 million cells | State Space Model (SSM) | Efficient alternative [62] |
Efficient Alternatives to Transformers
The quadratic complexity of standard transformer architectures has prompted development of more efficient alternatives:
State Space Models (SSMs): GeneMamba employs a BiMamba module to capture gene context information with linear computational complexity, significantly reducing resource requirements while maintaining competitive performance [62].
Retention-based Architectures: CellFM utilizes an ERetNet framework, a transformer variant with linear complexity, enabling training on 100 million cells with 800 million parameters [38].
Hybrid Approaches: Some models implement low-rank adaptation (LoRA) during fine-tuning to reduce trainable parameters when adapting to new datasets [38].
Integrated Decision Framework
Holistic Model Selection Strategy
Effective model selection requires simultaneous consideration of all three factors. Benchmarking studies recommend using a non-dominated sorting algorithm that aggregates multiple evaluation metrics to guide model selection [3]. Additionally, the roughness index (ROGI) can serve as a proxy to recommend appropriate models in a dataset-dependent manner [3].
Decision Workflow for Researchers
Experimental Protocols for Model Evaluation
When benchmarking scFMs for specific applications, researchers should implement the following standardized evaluation protocol:
Task Formulation: Clearly define the biological question and corresponding computational task (gene-level, cell-level, or spatial analysis) [3].
Baseline Establishment: Compare scFM performance against traditional methods appropriate for the task:
- Highly Variable Genes (HVGs) selection
- Anchor-based methods (Seurat)
- Clustering-based integration (Harmony)
- Generative models (scVI) [3]
Evaluation Metrics: Employ comprehensive metrics spanning unsupervised, supervised, and knowledge-based approaches:
Robustness Validation: Introduce independent, unbiased datasets (e.g., AIDA v2 from CellxGene) to mitigate data leakage risks and validate conclusions [3].
Table 4: Research Reagent Solutions for scFM Applications
| Resource Category | Specific Tools/Databases | Function and Application |
|---|---|---|
| Data Repositories | CELLxGENE, NCBI GEO, ENA, GSA, ImmPort [1] [38] | Source of standardized single-cell datasets for model training and validation |
| Unified Frameworks | BioLLM [23] | Standardized APIs for model integration and benchmarking across diverse scFMs |
| Processing Tools | SynEcoSys single-cell database [38] | Quality control, gene name standardization, and format unification |
| Benchmarking Datasets | Asian Immune Diversity Atlas (AIDA) v2 [3] [51] | Independent validation dataset to mitigate data leakage risks |
| Knowledge Bases | STRING database [63] | Source of protein-protein interactions for knowledge-enhanced models |
Selecting appropriate single-cell foundation models requires careful consideration of dataset characteristics, analytical tasks, and computational constraints. Evidence from comprehensive benchmarks indicates that while scFMs provide robust and versatile tools for diverse applications, simpler machine learning models may be more efficient for specific datasets, particularly under resource constraints [3]. The field is rapidly evolving with emerging trends including more efficient architectures like state space models [62], integration of multi-omics data [1], and development of spatially-aware models [8]. As these models continue to mature, they hold tremendous promise for advancing drug development and precision medicine by providing deeper insights into cellular function and disease mechanisms.
The advent of single-cell RNA sequencing (scRNA-seq) has revolutionized biology by allowing researchers to probe cellular heterogeneity at an unprecedented resolution. Concurrently, the field of artificial intelligence has witnessed the rise of foundation models—large-scale deep learning models pretrained on vast datasets that can be adapted to a wide range of downstream tasks [1]. The convergence of these two domains has given rise to single-cell foundation models (scFMs), which aim to learn universal representations of cellular biology from millions of single-cell transcriptomes [1] [3]. These models typically employ transformer architectures, originally developed for natural language processing, to interpret the "language" of cells, where genes are treated as words and entire cells as sentences [1]. The fundamental premise is that by exposing a model to massive and diverse single-cell datasets encompassing numerous tissues, species, and conditions, it can capture the fundamental principles governing cellular identity and function, which can then be generalized to new biological questions with minimal fine-tuning [1] [3].
This technical guide examines the core factors that determine the performance and longevity of scFMs, with a specific focus on model scale and data diversity. As the field rapidly evolves, understanding the relationship between these factors and model performance is crucial for developing robust, future-proof tools that can accelerate biological discovery and therapeutic development [3] [64]. We synthesize evidence from recent benchmarking studies, provide detailed experimental protocols for evaluating scFMs, and offer practical guidance for researchers seeking to leverage these powerful models in their work.
Architectural Foundations of Single-Cell Foundation Models
Core Model Architectures and Tokenization Strategies
Most scFMs are built on transformer architectures, which utilize attention mechanisms to weight the relationships between input tokens [1]. In natural language processing, this allows models to determine which words in a sentence are most important when predicting missing words. Similarly, in scFMs, attention mechanisms learn which genes in a cell are most informative about cellular identity and state, capturing how genes co-vary across cells and potentially reflect functional connections [1]. Two predominant architectural paradigms have emerged:
- Encoder-based models (e.g., scBERT): These employ bidirectional attention mechanisms that learn from all genes in a cell simultaneously, making them particularly suited for classification tasks and generating cell embeddings [1].
- Decoder-based models (e.g., scGPT): These use unidirectional masked self-attention that iteratively predicts masked genes conditioned on known genes, offering strengths in generative tasks [1].
A critical preprocessing step for all scFMs is tokenization—converting raw gene expression data into discrete units (tokens) that the model can process. Unlike words in a sentence, genes have no inherent ordering, presenting a unique challenge. Common tokenization strategies include [1]:
- Expression-based ranking: Genes are ordered by their expression levels within each cell, creating a deterministic sequence.
- Binning approaches: Genes are partitioned into bins based on expression values.
- Normalized counts: Some models report no clear advantage to complex ranking and simply use normalized expression values.
Table 1: Tokenization Strategies in Prominent Single-Cell Foundation Models
| Model | Tokenization Approach | Positional Encoding | Special Tokens |
|---|---|---|---|
| scGPT | Expression value bins + gene IDs | Learnable embeddings | Cell identity, modality, batch |
| Geneformer | Top highly-expressed genes | Rank-based encoding | None reported |
| scBERT | Expression bins | Fixed positional encoding | Cell type tokens |
| UCE | Normalized counts | Not specified | Tissue and species metadata |
After tokenization, genes are typically represented as embedding vectors that combine a gene identifier with its expression value. Positional encoding schemes are then applied to represent the relative order or rank of each gene in the cell [1]. Special tokens may be added to represent cell-level context, experimental modality, or batch information, enriching the model's understanding of the biological context [1].
The Scaling Laws: Model Size and Performance
The "foundation" in foundation models implies substantial scale, both in terms of architecture and training data. In natural language processing, large language models have demonstrated predictable scaling laws where performance improves with increased model size and training data [65]. Early evidence suggests similar relationships may exist in biological foundation models, though the field is still in its infancy relative to its NLP counterparts.
Model scale in scFMs encompasses multiple dimensions [1]:
- Parameter count: The number of trainable weights in the neural network architecture.
- Training data volume: The number of single cells used for pretraining.
- Data diversity: The breadth of biological conditions covered (cell types, tissues, species, experimental conditions).
The scaling potential of biological foundation models is exemplified by protein structure prediction models like AlphaFold, which demonstrated that increased model capacity coupled with diverse training data can solve long-standing biological challenges [65] [64]. For single-cell models, scaling is complicated by the heterogeneous nature of single-cell data, which exhibits high sparsity, high dimensionality, and significant technical noise [3]. Recent benchmarking studies have begun to systematically explore how scale affects performance across different biological tasks, with nuanced findings that question whether simply scaling up always yields better performance [3] [66].
Data Diversity: The Biological Corpus for Model Training
The performance and generalizability of scFMs are fundamentally constrained by the quality and diversity of their training data. Significant effort has been invested in curating large-scale single-cell atlases that provide comprehensive coverage of cell types and states. Key data sources for pretraining scFMs include [1]:
- CZ CELLxGENE: Provides unified access to annotated single-cell datasets, with over 100 million unique cells standardized for analysis.
- Human Cell Atlas: A global consortium building comprehensive reference maps of all human cells.
- Public repositories: NCBI GEO, SRA, and EMBL-EBI Expression Atlas host thousands of single-cell sequencing studies.
- Curated compendia: PanglaoDB and Human Ensemble Cell Atlas collate data from multiple sources and studies.
The assembly of a high-quality, non-redundant dataset for pretraining is considered as important as model architecture for building robust scFMs [1]. Critical challenges in data curation include handling batch effects, technical noise from different experimental protocols, varying sequencing depths, and inconsistent processing steps across studies [1]. Effective pretraining requires careful selection of datasets, filtering of cells and genes, balancing dataset compositions, and rigorous quality control [1].
The Impact of Data Diversity on Model Generalization
Data diversity serves as a regularization mechanism that prevents models from overfitting to technical artifacts or specific biological contexts. A comprehensive benchmark study evaluated six scFMs against established baselines across multiple tasks and found that data diversity during pretraining significantly impacts performance on challenging biological problems [3]. The study demonstrated that models trained on more diverse datasets showed improved performance on tasks including:
- Batch integration: Harmonizing datasets from different experimental batches while preserving biological variation.
- Cell type annotation: Identifying and labeling cell types in new datasets.
- Cancer cell identification: Distinguishing malignant cells in tumor microenvironments.
- Drug sensitivity prediction: Forecasting cellular responses to therapeutic compounds.
The benchmarking revealed that scFMs excel particularly in zero-shot learning scenarios, where models must perform tasks without task-specific fine-tuning, suggesting that diverse pretraining enables the acquisition of fundamental biological principles [3]. This capability is crucial for applications in drug discovery, where models must often predict effects for novel compounds or in understudied cell types [64].
Quantitative Benchmarking: Evaluating the Impact of Scale and Diversity
Benchmarking Methodology and Metrics
Rigorous benchmarking is essential for quantifying the relationship between model scale, data diversity, and performance. Recent studies have developed comprehensive evaluation frameworks that assess scFMs across multiple biological tasks using both traditional metrics and novel biologically-grounded approaches [3]. Key evaluation dimensions include:
- Gene-level tasks: Assessing whether functionally similar genes are embedded close together in the latent space, evaluated through gene ontology term prediction and tissue specificity analysis.
- Cell-level tasks: Evaluating cell type annotation accuracy, batch integration capability, and compositional analysis across conditions.
- Perturbation prediction: Measuring accuracy in forecasting cellular responses to genetic or chemical perturbations.
Innovative evaluation metrics introduced in recent benchmarks include [3]:
- scGraph-OntoRWR: Measures the consistency of cell type relationships captured by scFMs with prior biological knowledge from cell ontologies.
- Lowest Common Ancestor Distance (LCAD): Assesses the severity of errors in cell type annotation by measuring the ontological proximity between misclassified cell types.
- Roughness Index (ROGI): Quantifies the smoothness of the cell-property landscape in the latent space, with smoother landscapes correlating with better downstream task performance.
Table 2: Performance of Models on Key Biological Tasks (Pearson Correlation in Differential Expression Space)
| Model | Adamson Dataset | Norman Dataset | Replogle K562 | Replogle RPE1 | Batch Integration | Cell Type Annotation |
|---|---|---|---|---|---|---|
| scGPT | 0.641 | 0.554 | 0.327 | 0.596 | 0.712 | 0.685 |
| scFoundation | 0.552 | 0.459 | 0.269 | 0.471 | 0.698 | 0.662 |
| Geneformer | N/A | N/A | N/A | N/A | 0.721 | 0.694 |
| Random Forest (GO features) | 0.739 | 0.586 | 0.480 | 0.648 | N/A | N/A |
| Train Mean (baseline) | 0.711 | 0.557 | 0.373 | 0.628 | N/A | N/A |
Surprising Limitations of Current Scaling Approaches
Despite the theoretical benefits of scale, recent benchmarking studies have revealed unexpected limitations in current scFMs. A systematic evaluation of perturbation prediction capabilities found that foundation models underperformed simpler baselines across multiple datasets [66]. Surprisingly, even the simplest baseline model—which predicts the average expression profile from training data—outperformed both scGPT and scFoundation on standard Perturb-seq benchmarks [66]. Furthermore, basic machine learning models incorporating biologically meaningful features (e.g., Gene Ontology vectors) significantly outperformed foundation models [66].
These findings suggest that current scaling approaches may not be efficiently translating into improved performance for specific tasks. Potential explanations include [66]:
- Low perturbation-specific variance in benchmark datasets, making them suboptimal for evaluating complex models.
- Inefficient knowledge extraction from pretraining, where models fail to capture biologically relevant gene-gene relationships.
- Architectural mismatches between the pretraining objectives and downstream tasks.
However, it's noteworthy that using scFM-generated embeddings as features in traditional machine learning models improved performance compared to the end-to-end fine-tuned foundation models themselves, suggesting that these models do capture useful biological information that isn't fully leveraged by their native architectures [66].
Experimental Protocols for Evaluating Scale and Diversity
Protocol 1: Assessing Data Diversity Effects
Objective: Quantify the impact of training data diversity on model generalizability across tissue types and species.
Materials:
- Single-cell datasets: Curate datasets from diverse sources including CELLxGENE, Human Cell Atlas, and GEO.
- Computing infrastructure: High-performance computing cluster with multiple GPUs (e.g., NVIDIA A100 or H100).
- Software: Python with scFMs implementations (scGPT, Geneformer, scFoundation).
Methodology:
- Data stratification: Partition pretraining data along diversity axes (tissue types, species, experimental protocols).
- Model training: Train identical model architectures on datasets with varying diversity levels.
- Zero-shot evaluation: Assess performance on held-out tissues/cell types without fine-tuning.
- Fine-tuning evaluation: Measure sample efficiency when adapting to new tasks with limited labeled data.
Analysis:
- Calculate performance metrics (accuracy, F1-score) across diversity conditions.
- Use scGraph-OntoRWR to quantify biological consistency of embeddings.
- Apply ROGI to measure landscape smoothness for different training regimes.
Protocol 2: Benchmarking Perturbation Prediction
Objective: Evaluate how model scale affects prediction of cellular responses to genetic and chemical perturbations.
Materials:
- Perturbation datasets: Adamson, Norman, and Replogle Perturb-seq datasets.
- Baseline models: Implement Train Mean, k-NN, Random Forest, and ElasticNet regressors.
- Evaluation framework: Standardized metrics for pseudo-bulk and differential expression correlation.
Methodology:
- Data preprocessing: Normalize counts and create pseudo-bulk profiles for each perturbation.
- Model configuration: Test foundation models at varying scales (parameter counts).
- Feature extraction: Compare foundation model embeddings against traditional biological features (Gene Ontology, pathway databases).
- Performance assessment: Evaluate using Pearson correlation in differential expression space.
Analysis:
- Compare performance across model scales and architectural variants.
- Identify specific perturbation types where scale provides maximal benefit.
- Analyze embedding spaces to determine whether larger models capture more biologically meaningful relationships.
Diagram 1: Experimental Workflow for Evaluating scFMs
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for Single-Cell Foundation Model Research
| Reagent/Resource | Type | Function | Example Sources/Implementations |
|---|---|---|---|
| CELLxGENE Database | Data Resource | Provides standardized, annotated single-cell data for pretraining and benchmarking | CZ CELLxGENE (>100M cells) [1] |
| Perturb-seq Datasets | Benchmark Data | Enables evaluation of perturbation prediction capabilities | Adamson, Norman, Replogle datasets [66] |
| Gene Ontology Annotations | Biological Prior Knowledge | Provides ground truth for evaluating biological relevance of gene embeddings | Gene Ontology Consortium [3] |
| scGraph-OntoRWR | Evaluation Metric | Quantifies consistency of model-derived cell relationships with ontological knowledge | Custom implementation [3] |
| Pretrained Model Weights | Model Resource | Enables transfer learning without expensive pretraining | scGPT, Geneformer, scFoundation [1] [66] |
Path Forward for Scalable and Robust Single-Cell Foundation Models
The field of single-cell foundation models stands at a critical juncture, where the initial promise of large-scale models must be reconciled with nuanced benchmarking results that show inconsistent advantages over simpler approaches [3] [66]. Future progress will likely depend on several key developments:
- Improved pretraining objectives: Designing self-supervised tasks that better capture causal biological relationships rather than correlational patterns.
- Multimodal integration: Incorporating additional data modalities beyond transcriptomics, such as epigenetics (scATAC-seq), proteomics, and spatial information to create more comprehensive cellular representations [1].
- Architectural innovations: Developing transformer variants specifically optimized for the unique characteristics of biological data, which lacks inherent sequence unlike language.
- Interpretability advances: Creating methods to extract biologically meaningful insights from the latent representations learned by these models [1].
The commercial applications of scFMs in drug discovery continue to advance, with companies leveraging these models to identify novel therapeutic targets, design optimized biologics, and predict compound efficacy and toxicity [65] [64]. The emerging paradigm involves using publicly available foundation models as a starting point, which are then fine-tuned on proprietary datasets to address specific therapeutic questions [64]. This approach significantly lowers computational costs while allowing organizations to maximize the value of their unique data assets.
Based on current evidence, future-proofing scFM development requires a balanced approach that considers both scale and efficiency. Rather than indiscriminately scaling model size and training data, researchers should:
- Prioritize data quality and biological diversity over sheer volume of cells.
- Invest in comprehensive benchmarking across multiple biological tasks to identify failure modes and improvement opportunities.
- Develop task-specific model selection criteria rather than seeking universal dominance from any single architecture.
- Focus on interpretability to build trust and facilitate biological discovery from model representations.
The trajectory of scFMs suggests they will become increasingly central to single-cell research and drug development, but their evolution must be guided by rigorous empirical evaluation rather than scaling for its own sake. By strategically focusing on both model scale and data diversity while maintaining connection to biological plausibility, the next generation of scFMs promises to unlock deeper insights into cellular function and disease mechanisms, ultimately accelerating the development of novel therapeutics.
Benchmarking Reality: How scFMs Stack Up Against Traditional Methods
Single-cell foundation models (scFMs) represent a transformative advancement in computational biology, applying large-scale deep learning to single-cell RNA sequencing (scRNA-seq) data. Inspired by the success of foundation models in natural language processing (NLP), these models are trained on millions of single-cell transcriptomes to learn fundamental biological principles that can be adapted to various downstream tasks [1]. The development of scFMs addresses critical challenges in single-cell data analysis, including the high sparsity, dimensionality, and technical noise inherent in scRNA-seq data [3]. By leveraging self-supervised learning on massive datasets, scFMs capture universal patterns of gene expression and cellular behavior, enabling researchers to extract meaningful biological insights from complex cellular landscapes [1] [60]. This whitepaper presents a comprehensive benchmarking study of six leading scFMs, evaluating their performance across diverse biological tasks to guide researchers and drug development professionals in selecting appropriate models for specific applications.
Methodology: Benchmarking Framework and Experimental Design
Our benchmarking framework evaluates six prominent scFMs (Geneformer, scGPT, UCE, scFoundation, LangCell, and scCello) against well-established baseline methods under realistic conditions [3]. The evaluation encompasses two gene-level tasks (tissue specificity prediction and Gene Ontology term prediction) and four cell-level tasks (batch integration, cell type annotation, cancer cell identification, and drug sensitivity prediction) [3]. Model performance is assessed using 12 metrics spanning unsupervised, supervised, and knowledge-based approaches, including novel biologically informed metrics such as scGraph-OntoRWR and Lowest Common Ancestor Distance (LCAD) [3].
Selected Models and Baseline Methods
Table 1: Overview of Benchmarked Single-Cell Foundation Models
| Model Name | Architecture Type | Pretraining Data Scale | Key Features |
|---|---|---|---|
| Geneformer | Transformer-based | 30 million cells [67] | Encoder-based architecture; rank-based gene ordering |
| scGPT | Transformer-based | Not specified | Decoder-based architecture; multimodal capability |
| scFoundation | Transformer-based | 100 million cells [6] | Large-scale pretraining on diverse cell types |
| UCE | Transformer-based | Not specified | Unified cell embedding |
| LangCell | Transformer-based | Not specified | Natural language processing inspired |
| scCello | Transformer-based | Not specified | Focus on cellular dynamics |
| scBERT | Transformer-based | Not specified | Bidirectional encoder; masked language modeling |
Baseline methods included traditional approaches such as Highly Variable Genes (HVGs) selection, anchor-based Seurat, clustering-based Harmony, and the generative model scVI [3]. These baselines provide reference points for evaluating whether the complex pretraining of scFMs offers tangible advantages over established methods.
Datasets and Evaluation Metrics
The benchmark utilizes diverse datasets with high-quality labels, including five datasets for preclinical batch integration and cell type annotation, and seven cancer types with four drugs for clinically relevant tasks [3]. To mitigate data leakage concerns and validate conclusions rigorously, an independent and unbiased dataset—the Asian Immune Diversity Atlas (AIDA) v2 from CellxGene—was introduced [3].
Performance was evaluated using multiple metrics, including:
- Average Silhouette Width (ASW) for cluster separation quality [60]
- scGraph-OntoRWR for consistency with biological knowledge [3]
- Lowest Common Ancestor Distance (LCAD) for ontological proximity of misclassifications [3]
- Standard classification metrics (accuracy, F1-score) for supervised tasks [60]
Experimental Workflow
The following diagram illustrates the comprehensive benchmarking workflow, from data input through task evaluation:
Diagram 1: Benchmarking Workflow for scFM Evaluation
Results: Performance Analysis Across Tasks
Gene-Level Task Performance
Gene-level tasks evaluate how well scFMs capture functional relationships between genes, which is crucial for understanding biological mechanisms. In these tasks, models extract gene embeddings from their input layers and use them to predict known biological relationships, including tissue specificity and Gene Ontology terms [3].
Table 2: Performance Comparison on Gene-Level Tasks
| Model | Tissue Specificity Prediction | GO Term Prediction | Notable Strengths |
|---|---|---|---|
| Geneformer | High | High | Effective pretraining strategy for gene relationships |
| scGPT | Moderate | High | Robust across multiple task types |
| scFoundation | High | High | Benefits from large-scale pretraining |
| UCE | Moderate | Moderate | Balanced performance |
| scBERT | Lower | Lower | Limited by model size and training data |
Our findings indicate that Geneformer and scFoundation demonstrate particularly strong capabilities in gene-level tasks, benefiting from effective pretraining strategies that effectively capture gene-gene relationships [23] [60]. These models successfully embed functionally similar genes in close proximity within the latent space, analogous to how words with similar meanings cluster in NLP models [3].
Cell-Level Task Performance
Batch Integration and Cell Type Annotation
Batch integration evaluates how well models can remove technical artifacts while preserving biological variation, which is crucial for building unified cell atlases [3]. Cell type annotation assesses the models' ability to correctly identify cell types, a fundamental task in single-cell analysis.
In zero-shot settings, scGPT consistently outperformed other models in generating biologically relevant cell embeddings, achieving superior separation of cell types as visualized through UMAP projections [60]. The model demonstrated particular effectiveness in preserving biologically relevant information, making it more effective for clustering tasks [60].
For batch-effect-removal capabilities assessed using joint datasets with varying degrees of batch effects, scGPT again outperformed other models across metrics, yielding superior results compared to principal component analysis (PCA) [60]. Other models generally performed worse than PCA in this task [60].
Clinical Application Tasks
For clinically relevant tasks including cancer cell identification and drug sensitivity prediction, performance varied significantly across models and cancer types. The benchmarking revealed that no single scFM consistently outperformed others across all tasks, emphasizing the need for task-specific model selection [3].
Notably, fine-tuning through supervised training significantly enhanced performance for both cell embedding extraction and batch-effect correction [60]. This highlights the importance of incorporating task-specific fine-tuning to optimize the accuracy and reliability of cell embeddings for clinical applications.
Impact of Input Sequence Length and Computational Efficiency
We investigated how the number of input genes affected embedding quality across models. scGPT's embeddings became more accurate with longer input sequences, suggesting that richer information enables better cell representations [60]. In contrast, Geneformer and scFoundation showed slight negative correlations in some datasets, while scBERT's performance declined with longer sequences across most datasets [60].
Computational resource assessment revealed that scGPT and Geneformer demonstrated superior efficiency in memory usage and computational time compared to scBERT and scFoundation, underscoring their practicality for large-scale analyses [60].
Technical Protocols: Implementation Guidelines
Model Training and Fine-tuning Procedures
The benchmarking studies employed both zero-shot evaluation and fine-tuning approaches. For zero-shot evaluation, models were used without additional training on downstream tasks, using pretrained embeddings directly for analysis [3] [60]. For fine-tuning, models were further trained on specific tasks with labeled data, which significantly enhanced performance metrics [60] [67].
The closed-loop fine-tuning approach, which incorporates experimental perturbation data during model refinement, has shown particular promise. This method increased positive predictive value three-fold—from 3% to 9%—with concurrent improvements in negative predictive value (99%), sensitivity (76%), and specificity (81%) in T-cell activation studies [67].
Interpretation and Biological Validation Methods
To enhance interpretability, researchers have developed novel approaches such as transcoder-based circuit analysis, which extracts internal decision-making circuits from scFMs [68]. This method trains transcoders on scFMs to decompose model computations into interpretable components, establishing correspondences between extracted circuit components and biological knowledge [68].
Additionally, biologically informed metrics such as scGraph-OntoRWR measure the consistency of cell type relationships captured by scFMs with prior biological knowledge, while the Lowest Common Ancestor Distance (LCAD) metric assesses the severity of error in cell type annotation by measuring ontological proximity between misclassified cell types [3].
Computational Frameworks and Benchmarking Tools
Table 3: Essential Computational Resources for scFM Research
| Resource Name | Type | Function | Application |
|---|---|---|---|
| BioLLM | Unified framework | Standardizes deployment of scFMs through integrated modules | Enables seamless model switching and comparative evaluation [23] [60] |
| CellxGene | Data platform | Provides unified access to annotated single-cell datasets | Source of standardized data for training and validation [3] [1] |
| SpatialCorpus-110M | Curated data resource | One of the largest collections of single-cell and spatial data | Enables spatial context modeling in foundation models [8] |
| Transcoder | Interpretability tool | Extracts internal decision circuits from neural networks | Provides biological interpretation of model predictions [68] |
Experimental Design Considerations
Based on comprehensive benchmarking results, we recommend the following guidelines for researchers selecting scFMs:
- For gene-level tasks: Prioritize Geneformer or scFoundation due to their effective pretraining strategies for capturing gene-gene relationships [23] [60]
- For cell-level tasks: Consider scGPT for its robust performance across multiple cell-level applications, particularly in zero-shot settings [60]
- For resource-constrained environments: Simpler machine learning models may be more adept at efficiently adapting to specific datasets, particularly under limited computational resources [3]
- For specialized applications: Consider domain-specific models like scPlantLLM for plant single-cell genomics [6] or Nicheformer for spatial context modeling [8]
Comprehensive benchmarking of six leading single-cell foundation models reveals a nuanced landscape where model performance is highly task-dependent. While scFMs demonstrate robust capabilities as versatile tools for diverse applications, simpler machine learning models can be more efficient for specific datasets, particularly under resource constraints [3]. Notably, no single scFM consistently outperforms others across all tasks, emphasizing the need for tailored model selection based on factors such as dataset size, task complexity, biological interpretability requirements, and computational resources [3].
The introduction of novel evaluation perspectives, including biologically informed metrics and clinically relevant tasks, provides deeper insights into the strengths and limitations of current scFMs [3]. The development of standardized frameworks like BioLLM further enhances the accessibility and reproducibility of scFM research [23] [60].
Future directions in scFM development include enhanced spatial context modeling [8], improved interpretability methods [68], closed-loop frameworks that incorporate experimental data [67], and domain-specific adaptations for specialized applications [6]. As these models continue to evolve, they hold tremendous promise for advancing biological discovery and therapeutic development through more accurate and interpretable computational representations of cellular behavior.
As the field progresses toward the vision of comprehensive "virtual cell" models, systematic benchmarking and standardized evaluation will remain crucial for guiding model selection and development, ultimately accelerating our understanding of cellular function in health and disease.
Single-cell RNA sequencing (scRNA-seq) has revolutionized biological research by enabling high-resolution analysis of gene expression at the individual cell level, uncovering cellular heterogeneity, developmental trajectories, and complex regulatory networks [1] [6]. The exponential growth of single-cell data has created both an opportunity and a pressing need for computational methods capable of integrating and extracting universal biological patterns from these vast datasets. Inspired by breakthroughs in natural language processing (NLP), researchers have developed single-cell foundation models (scFMs)—large-scale deep learning models pretrained on massive single-cell atlases that can be adapted to diverse downstream tasks [1] [2].
These models, including prominent examples such as scGPT, Geneformer, and scBERT, conceptualize cellular data in linguistic terms: cells are treated as "sentences" and genes or genomic features as "words" [1]. Through self-supervised pretraining on millions of cells, scFMs aim to learn fundamental biological principles that enable zero-shot application or efficient fine-tuning for specific analyses like cell type annotation, batch integration, and perturbation prediction [1] [51].
Despite their promising potential, rigorous benchmarking studies have raised critical questions about the performance of these emerging scFMs relative to established traditional methods. This whitepaper provides a comprehensive technical comparison of three leading scFMs—scGPT, Geneformer, and scBERT—against simpler, well-established computational approaches, evaluating their capabilities across fundamental single-cell analysis tasks to guide researchers and drug development professionals in selecting appropriate tools for their specific applications.
Core Architectural Foundations
Model Architectures and Pretraining Approaches
Single-cell foundation models share a common conceptual foundation but diverge significantly in their architectural implementations and pretraining strategies. Most scFMs are built on the transformer architecture, which utilizes attention mechanisms to model relationships between genes, but they differ in their specific configurations and training objectives [1].
scGPT employs a decoder-based architecture inspired by the Generative Pretrained Transformer (GPT), using a unidirectional masked self-attention mechanism that iteratively predicts masked genes conditioned on known genes [1] [69]. It incorporates both gene identity and expression value embeddings, with expression values typically binned into discrete ranges. scGPT is pretrained on diverse datasets, including 33 million non-cancerous human cells from various tissues, using iterative masked gene modeling with mean square error loss [51] [69].
Geneformer utilizes an encoder-based architecture with bidirectional attention, allowing it to learn from all genes in a cell simultaneously [69]. Rather than binning expression values, Geneformer employs a unique ranking system where genes are ordered by expression level within each cell, using positional encoding to embed expression information [69]. It was pretrained on approximately 30 million cells using masked gene modeling with cross-entropy loss focused on gene identity prediction [51] [69].
scBERT follows a BERT-like encoder architecture with bidirectional attention mechanisms [1] [69]. Similar to scGPT, it incorporates both gene identity and binned expression value embeddings. scBERT was pretrained on a comparatively smaller dataset of over 1.1 million cells from PanglaoDB, using masked language modeling objectives [69].
The table below summarizes key architectural differences:
Table 1: Architectural Comparison of Single-Cell Foundation Models
| Model | Architecture Type | Parameters | Pretraining Dataset Size | Input Representation | Value Embedding | Positional Embedding |
|---|---|---|---|---|---|---|
| scGPT | Decoder-based Transformer | ~50 million | 33 million cells | 1200 highly variable genes | Value binning | No |
| Geneformer | Encoder-based Transformer | ~40 million | 30 million cells | 2048 ranked genes | Expression ranking | Yes |
| scBERT | Encoder-based Transformer (BERT) | Not specified | 1.1 million cells | Gene ranking with bins | Value binning | Not specified |
Tokenization Strategies for Single-Cell Data
A critical challenge in adapting transformer architectures to single-cell data is the non-sequential nature of gene expression, unlike natural language where word order carries semantic meaning [1] [3]. scFMs address this through various tokenization strategies that convert raw gene expression data into structured model inputs.
The tokenization process typically involves three components: gene embeddings (representing gene identity), value embeddings (representing expression levels), and positional embeddings (providing sequence context) [51]. Models employ different strategies to impose order on inherently unordered gene expression data. scGPT typically uses highly variable genes without complex ordering, while Geneformer and some other models rank genes by expression level within each cell to create a deterministic sequence [1] [69]. Expression values are commonly handled through binning into discrete ranges or using normalized counts [1].
Additional special tokens may be incorporated to enrich biological context, including cell identity tokens, modality indicators for multi-omics data, and batch information tokens [1]. These tokens are converted to embedding vectors processed by transformer layers, ultimately producing latent embeddings at both the gene and cell levels that capture biological relationships and patterns [1].
Diagram 1: Architectural comparison of scFMs showing divergent strategies despite shared transformer foundation
Experimental Protocols for Benchmarking
Standardized Evaluation Framework
Rigorous benchmarking of scFMs requires standardized experimental protocols that assess model performance across diverse tasks and datasets. Comprehensive evaluations typically employ zero-shot settings where pretrained models generate embeddings without task-specific fine-tuning, as well as fine-tuning paradigms that adapt models to specific downstream applications [55] [51]. These protocols utilize multiple high-quality datasets with manual annotations that vary in size, complexity, and biological diversity, incorporating batch effects from different sources including inter-patient, inter-platform, and inter-tissue variations [51].
Evaluation metrics span unsupervised, supervised, and knowledge-based approaches. Traditional metrics include Average BIO (AvgBio) score for cell type clustering and average silhouette width (ASW) for cluster separation [55]. More recently, biologically-informed metrics such as scGraph-OntoRWR have been developed to measure the consistency of cell type relationships captured by scFMs with established biological knowledge from cell ontologies [51] [3]. The Lowest Common Ancestor Distance (LCAD) metric assesses the severity of cell type misclassification by measuring ontological proximity between incorrectly assigned types [51].
Benchmarking studies typically compare scFMs against established traditional methods including:
- Selection of highly variable genes (HVG)
- Harmony, a clustering-based integration method [55] [51]
- scVI, a generative model for single-cell data [55] [51]
- Seurat, an anchor-based integration approach [51]
Key Benchmarking Tasks and Datasets
Performance evaluation encompasses both gene-level and cell-level tasks. Gene-level tasks assess the biological relevance of learned gene embeddings by evaluating their ability to predict functional relationships, tissue specificity, and Gene Ontology terms [51] [3]. Cell-level tasks focus on practical applications including cell type annotation, batch integration, and identification of novel cell types [51].
Commonly used benchmark datasets include:
- Pancreas dataset: Combines data from five different sources with significant technical variation [55]
- PBMC (12k) dataset: Peripheral blood mononuclear cells with relatively low complexity [55]
- Tabula Sapiens: A comprehensive multi-organ atlas with diverse cell types [55]
- Immune datasets: Feature various immune cell types across different conditions [55]
- Asian Immune Diversity Atlas (AIDA) v2: Used for independent validation to mitigate data leakage concerns [51]
For perturbation prediction tasks, specialized datasets such as the Norman et al. CRISPR activation data (covering 100 individual genes and 124 gene pairs) and Replogle et al. CRISPR interference datasets provide ground truth for evaluating model ability to predict transcriptome changes after genetic perturbations [70].
Diagram 2: Comprehensive benchmarking workflow for evaluating scFMs across multiple tasks and metrics
Performance Comparison Across Key Tasks
Cell Type Annotation and Clustering
Cell type identification represents a fundamental application of single-cell analysis, where foundation models aim to project noisy gene expression measurements into biologically relevant latent spaces that separate known cell types [55]. Evaluation of zero-shot performance in separating known cell types across multiple datasets reveals significant limitations in current scFMs.
In comprehensive benchmarks, both scGPT and Geneformer generally underperform simpler methods including highly variable gene selection and established approaches like Harmony and scVI when measured by Average BIO score and average silhouette width [55]. scGPT demonstrates variable performance across datasets, performing competitively on PBMC (12k) datasets but underperforming on others such as Tabula Sapiens and Immune datasets [55]. Geneformer consistently lags behind other methods across most evaluation metrics and datasets [55].
The table below summarizes quantitative performance comparisons for cell type annotation:
Table 2: Cell Type Annotation Performance Comparison (Zero-Shot)
| Method | Overall Ranking | Performance on PBMC | Performance on Tabula Sapiens | Performance on Immune Datasets | Key Strengths |
|---|---|---|---|---|---|
| HVG Selection | Top performer | Consistently high | Consistently high | Consistently high | Simplicity, reliability |
| Harmony | Competitive | Strong | Variable | Strong | Batch effect correction |
| scVI | Competitive | Strong | Strong | Variable | Generative modeling |
| scGPT | Variable | Strong | Moderate | Moderate | Tissue-specific adaptation |
| Geneformer | Underperforms | Weak | Weak | Weak | Network biology insights |
| scBERT | Underperforms | Weak | Weak | Weak | Limited testing |
Notably, pretraining provides clear benefits for scGPT, with models pretrained on specific tissues (e.g., blood and bone marrow) showing improved performance on related cell types [55]. However, the relationship between pretraining dataset size and cell type clustering performance appears nonlinear, with larger and more diverse datasets not consistently conferring additional benefits [55].
Batch Integration Capabilities
Batch integration represents a critical challenge in single-cell analysis, requiring the removal of technical artifacts from multiple data sources while preserving meaningful biological variation [55]. Evaluation of foundation models for this task reveals distinct strengths and limitations across different types of batch effects.
Visualization of embeddings on benchmark datasets like the Pancreas dataset (incorporating data from five different sources) shows that Geneformer largely fails to retain cell type information, with clustering primarily driven by batch effects [55]. scGPT provides better separation between cell types but still exhibits batch-effect-driven structure in dimensionality reductions [55]. In contrast, established methods like Harmony and scVI demonstrate more effective integration, successfully correcting for technical variations while preserving biological signals [55].
Quantitative evaluation with batch integration metrics confirms these observations, with Geneformer underperforming across most datasets and scGPT showing competitive performance specifically on complex datasets where both technical and biological batch effects are present [55]. Surprisingly, simple selection of highly variable genes achieves the best batch integration scores across all datasets when measured in full dimensions [55].
Perturbation Effect Prediction
Prediction of gene expression changes following genetic perturbations represents a promising application where foundation models could theoretically leverage pretrained knowledge of gene regulatory relationships. However, rigorous benchmarking against deliberately simple baselines reveals significant limitations in current capabilities [70].
In evaluations using CRISPR activation and interference datasets, foundation models including scGPT and scFoundation fail to outperform simple linear baselines for predicting transcriptome changes after single or double gene perturbations [70]. The "additive" baseline model, which simply sums the individual logarithmic fold changes of single perturbations to predict double perturbation effects, consistently outperforms all deep learning models [70]. Similarly, for predicting unseen perturbations, foundation models cannot consistently outperform the simple approach of predicting the mean expression across training perturbations [70].
When examining the ability to predict genetic interactions (where double perturbation effects deviate from additive expectations), no foundation model improves upon the "no change" baseline that always predicts control condition expression [70]. Furthermore, models show systematic biases in interaction type prediction, predominantly forecasting buffering interactions while rarely correctly predicting synergistic effects [70].
The Scientist's Toolkit: Essential Research Reagents
Implementing and evaluating single-cell foundation models requires specific computational resources and datasets. The table below details key "research reagent solutions" essential for working with scFMs:
Table 3: Essential Research Reagents for Single-Cell Foundation Model Research
| Resource Category | Specific Examples | Function/Purpose | Key Considerations |
|---|---|---|---|
| Pretrained Models | scGPT, Geneformer, scBERT weights | Provide starting point for transfer learning without costly pretraining | Model compatibility with data formats; licensing restrictions |
| Benchmark Datasets | Pancreas, PBMC, Tabula Sapiens, Immune datasets | Standardized evaluation across diverse biological contexts | Data quality; batch effect structure; cell type diversity |
| Evaluation Metrics | AvgBio, ASW, scGraph-OntoRWR, LCAD | Quantify performance across different task dimensions | Biological relevance; statistical robustness |
| Integration Frameworks | BioLLM, scEval | Standardized APIs for model comparison and application | Architecture compatibility; documentation quality |
| Computational Resources | GPU clusters (A100 or higher), 40+ GB memory | Enable model training and inference at scale | Cloud vs. local deployment; cost considerations |
| Data Repositories | CELLxGENE, PanglaoDB, GEO, SRA | Source of training data and evaluation benchmarks | Data standardization; metadata quality |
Notably, the BioLLM framework provides unified interfaces for diverse scFMs, addressing challenges posed by heterogeneous architectures and coding standards through standardized APIs and comprehensive documentation [23]. Similarly, the scEval platform offers reproducible benchmarking across multiple tasks and metrics, though it requires significant computational resources (GPU cores such as A100 and 40+ GB memory) [71].
Comprehensive benchmarking reveals a nuanced landscape for single-cell foundation models. While scFMs represent a promising architectural paradigm for learning universal patterns from massive single-cell atlases, their current practical utility is constrained by inconsistent performance across fundamental tasks [55] [51] [70].
The evidence indicates that no single foundation model consistently outperforms established traditional methods, with performance highly dependent on specific tasks and dataset characteristics [51]. scGPT demonstrates the most robust performance across diverse applications, particularly in zero-shot settings and fine-tuning paradigms [23]. Geneformer shows strengths in gene-level tasks and network biology applications but underperforms in basic cell type annotation and batch integration [55] [23]. scBERT generally lags behind other foundation models, likely due to smaller model size and limited training data [23].
For researchers and drug development professionals, selection between foundation models and traditional methods should be guided by specific application requirements, dataset characteristics, and computational resources. Simpler machine learning approaches often provide more efficient adaptation to specific datasets, particularly under resource constraints or when analyzing data similar to their training distribution [51]. Foundation models may offer advantages for exploratory analyses where labels are unknown and fine-tuning is impossible, or when leveraging their learned biological knowledge for hypothesis generation [55].
Future development should focus on improving pretraining objectives to better align with biological understanding, developing more effective adaptation techniques like parameter-efficient fine-tuning [69], and establishing standardized evaluation protocols that prioritize real-world biological relevance [51]. As these models continue to evolve, they hold tremendous potential for advancing single-cell genomics and unlocking deeper insights into cellular function and disease mechanisms—but realizing this potential requires honest assessment of current limitations and targeted addressing of existing performance gaps.
Single-cell foundation models (scFMs) have emerged as transformative tools in computational biology, leveraging large-scale single-cell transcriptomics data to learn universal representations of genes and cells [1]. Trained on tens of millions of cells through self-supervised learning objectives, these models promise to revolutionize everything from cell atlas construction to therapeutic discovery [51] [3]. However, as the field progresses, a critical question remains: how can we effectively evaluate whether these models capture biologically meaningful patterns rather than merely optimizing conventional computational metrics? Traditional evaluation approaches often rely on technical benchmarks that may not adequately assess a model's grasp of underlying biological principles.
The scGraph-OntoRWR metric represents a paradigm shift in evaluation methodology by directly measuring the alignment between model-derived cell relationships and established biological knowledge encoded in cell ontologies [51] [3] [72]. This approach moves beyond purely statistical measures to introduce a biologically-grounded assessment framework that evaluates whether the relational structure of cell types learned by scFMs reflects their known biological relationships. By incorporating prior biological knowledge directly into the evaluation process, scGraph-OntoRWR addresses a fundamental limitation in current benchmarking practices and provides a more nuanced understanding of what models actually learn about biology.
Understanding scGraph-OntoRWR: Conceptual Framework and Methodology
Theoretical Foundations
The scGraph-OntoRWR metric operates on the fundamental premise that a robust single-cell foundation model should organize cells in its latent space according to their actual biological relationships, not just technical similarities [3]. It evaluates whether the proximity and relationships between cell types in the model's embedding space align with their established positions in formal cell ontologies - structured, controlled vocabularies that capture known relationships between cell types based on developmental lineage, functional characteristics, and molecular signatures [72].
This approach addresses a critical gap in traditional evaluation methods, which often focus solely on quantitative performance metrics without assessing biological plausibility. As noted in benchmark studies, "the scGraph-OntoRWR metric measures the consistency of cell type relationships captured by scFMs with prior biological knowledge" [51]. This is particularly important because models might achieve high scores on technical benchmarks through overfitting or exploiting dataset-specific artifacts rather than genuinely understanding biological principles.
Methodological Workflow
The scGraph-OntoRWR methodology integrates computational modeling with biological knowledge representation through a multi-stage process. The following diagram illustrates the complete workflow from single-cell data input to metric calculation:
The computational workflow begins with the generation of cell embeddings from the foundation model, typically using zero-shot representations without task-specific fine-tuning [51] [3]. These embeddings are used to construct a k-nearest neighbor graph representing cell-cell relationships as captured by the model. In parallel, a biological reference graph is constructed from formal cell ontologies, where nodes represent cell types and edges represent known biological relationships (e.g., developmental lineage, functional similarity).
The core of the method applies Random Walk with Restart (RWR) algorithms to both graphs, simulating the propagation of similarity through the networks [51]. RWR is particularly well-suited for this task as it captures both direct and indirect relationships between cell types, reflecting the multi-scale organization of biological systems. The resulting similarity matrices from both the model-derived graph and the ontology-based graph are then compared using correlation measures, with higher correlations indicating better alignment between the model's representations and biological ground truth.
Experimental Framework and Benchmarking Protocol
Model Selection and Task Design
In the comprehensive benchmark study that introduced scGraph-OntoRWR, researchers evaluated six prominent single-cell foundation models against established baseline methods [51] [3]. The selected models represented diverse architectural approaches and pretraining strategies, including Geneformer, scGPT, UCE, scFoundation, LangCell, and scCello. These models were evaluated alongside traditional methods such as highly variable genes (HVGs) selection, Seurat, Harmony, and scVI to ascertain the specific gains attributable to large-scale pretraining.
The benchmarking protocol encompassed both gene-level and cell-level tasks to provide a holistic assessment of model capabilities [51] [3]. At the gene level, models were evaluated on tissue specificity prediction and Gene Ontology term prediction, assessing their ability to capture functional relationships between genes. At the cell level, evaluation included batch integration, cell type annotation, cancer cell identification, and drug sensitivity prediction. This multi-task approach ensured comprehensive assessment across diverse application scenarios relevant to both basic research and clinical translation.
Datasets and Evaluation Metrics
The benchmark utilized multiple high-quality datasets with manual annotations that varied in size and diversity, containing multiple sources of batch effects including inter-patient, inter-platform, and inter-tissue variations [3]. To mitigate the risk of data leakage and validate conclusions rigorously, researchers introduced an independent and unbiased dataset: the Asian Immune Diversity Atlas (AIDA) v2 from CellxGene [51]. This approach ensured that evaluations reflected real-world conditions and challenged models with novel cell types, cross-tissue homogeneity, and intra-tumor heterogeneity.
Performance was evaluated using 12 metrics spanning unsupervised, supervised, and knowledge-based approaches [51] [3]. Alongside scGraph-OntoRWR, researchers introduced the Lowest Common Ancestor Distance (LCAD) metric, which measures the ontological proximity between misclassified cell types to assess the severity of annotation errors. This ontology-informed perspective provided crucial insights that traditional computational metrics missed, enabling more biologically meaningful interpretation of model performance.
Key Experimental Findings
The benchmark results revealed several critical insights about single-cell foundation models and the utility of scGraph-OntoRWR [51] [3]:
No single scFM consistently outperformed others across all tasks, emphasizing that model selection must be tailored to specific applications, dataset characteristics, and available computational resources.
Foundation models demonstrated robust performance across diverse applications, but simpler machine learning models sometimes adapted more efficiently to specific datasets, particularly under resource constraints.
The pretrained zero-shot scFM embeddings captured meaningful biological insights into the relational structure of genes and cells, which proved beneficial for downstream tasks.
Performance improvements correlated with a "smoother landscape" in the pretrained latent space, reducing the difficulty of training task-specific models.
The following table summarizes the comparative performance of different models across key biological tasks as assessed in the benchmark study:
Table 1: Model Performance Ranking Across Biological Tasks [72]
| Model | Batch Integration | Cell Type Annotation | Cancer ID | Drug Sensitivity | Overall Ranking |
|---|---|---|---|---|---|
| Geneformer | 2 | 3 | 1 | 2 | 2 |
| scGPT | 3 | 2 | 3 | 3 | 3 |
| UCE | 1 | 4 | 4 | 4 | 4 |
| scFoundation | 4 | 1 | 2 | 1 | 1 |
| Traditional ML | 5 | 5 | 5 | 5 | 6 |
| HVG Selection | 6 | 6 | 6 | 6 | 5 |
The experimental results demonstrated that ontology-informed evaluation metrics like scGraph-OntoRWR provided crucial insights that traditional computational metrics missed [51] [72]. The metric proved particularly valuable for assessing the biological relevance of learned representations, revealing cases where models achieved high technical performance but organized cells in ways inconsistent with established biological knowledge.
Implementing scGraph-OntoRWR and related biologically-grounded evaluation metrics requires specific computational resources and biological knowledge bases. The following table outlines key components of the experimental framework:
Table 2: Essential Research Reagents and Resources for scGraph-OntoRWR Implementation [51] [3] [72]
| Reagent/Resource | Function | Biological Significance |
|---|---|---|
| Gene Embeddings | Numerical representations of genes in latent space | Capture functional similarities between genes based on co-expression patterns across diverse cellular contexts |
| Cell Ontologies | Structured vocabularies defining cell types and relationships | Provide biological ground truth for evaluating the relevance of model-derived cell relationships |
| Benchmark Datasets | Curated single-cell data with high-quality annotations | Enable standardized evaluation across diverse biological conditions and technical variations |
| Attention Mechanisms | Model components that identify important relationships between inputs | Reveal gene-gene interactions and regulatory relationships learned from data |
| GO Term Annotations | Gene Ontology functional classifications | Serve as biological prior knowledge for validating gene embeddings and functional predictions |
The integration of these resources enables a comprehensive evaluation framework that assesses not just technical performance but biological plausibility. Cell ontologies, in particular, provide the foundational knowledge structure that makes biologically-grounded assessment possible by formally defining cell types and their relationships based on developmental lineage, functional characteristics, and molecular signatures [72].
Implementation Protocol: Technical Specifications and Computational Requirements
Data Preprocessing and Normalization
The implementation of scGraph-OntoRWR begins with standardized data preprocessing to ensure consistent evaluation across models and datasets [51] [3]. Single-cell RNA sequencing data should undergo quality control to filter low-quality cells and genes, followed by normalization to account for technical variations in sequencing depth. Gene names must be standardized according to HUGO Gene Nomenclature Committee (HGNC) guidelines to ensure consistent mapping across datasets and ontology resources [38].
For the construction of the biological reference graph, cell type annotations should be mapped to established ontology frameworks such as the Cell Ontology (CL) or Uberon multi-species anatomy ontology [72]. This mapping enables the formal representation of relationships between cell types, including "isa" hierarchies (e.g., "CD4-positive T cell is a T cell") and "partof" relationships (e.g., "T cell is part of the immune system").
Computational Implementation
The following diagram illustrates the core algorithmic workflow for calculating the scGraph-OntoRWR metric, showing the parallel processing of model-derived and ontology-derived graphs:
The computational implementation requires specific parameter settings for the Random Walk with Restart algorithm. Based on the benchmark studies, recommended parameters include a restart probability between 0.7-0.8 and convergence threshold of 1e-6 [51]. The k-nearest neighbor graph for model-derived embeddings typically uses k=15-30, with exact values potentially optimized for specific dataset sizes and sparsity patterns.
For the similarity matrix comparison, Pearson correlation typically provides a robust measure of alignment between model-derived and ontology-derived structures [51]. The final scGraph-OntoRWR score represents the degree of concordance between the computational model's organization of cells and the biological ground truth, with higher scores indicating better preservation of known biological relationships.
Future Directions and Clinical Applications
The development of biologically-grounded evaluation metrics like scGraph-OntoRWR represents a significant advancement toward more meaningful assessment of single-cell foundation models. As the field progresses, these metrics are likely to evolve in several important directions. Future iterations may incorporate more dynamic aspects of biological systems, such as temporal developmental trajectories and response to perturbations [1] [3]. There is also growing interest in extending these approaches to multi-modal data integration, assessing how well models capture relationships across transcriptomic, epigenomic, proteomic, and spatial dimensions [73].
In clinical and translational applications, biologically-grounded assessment is particularly valuable for ensuring that models will generalize reliably to new patient populations and disease contexts [51] [3]. The benchmark study highlighted applications in cancer cell identification, tumor microenvironment characterization, and drug sensitivity prediction - all areas where biological plausibility is essential for clinical translation. As single-cell technologies continue to advance and datasets expand, metrics like scGraph-OntoRWR will play an increasingly important role in guiding the development of more robust, biologically-informed models that can truly advance our understanding of cellular function and disease mechanisms.
Single-cell RNA sequencing (scRNA-seq) has fundamentally transformed biological research by enabling the profiling of gene expression at the resolution of individual cells. This technology has revealed unprecedented insights into cellular heterogeneity, development, and disease mechanisms. However, the high-dimensionality, sparsity, and technical noise inherent in scRNA-seq data present significant computational challenges. Single-cell foundation models (scFMs) have emerged as a powerful class of computational tools designed to address these challenges. These large-scale deep learning models are pretrained on vast datasets comprising millions of cells and can be adapted for diverse downstream tasks. This whitepaper provides a technical examination of scFM performance across three critical analytical domains: automated cell type annotation, batch integration, and clinical prediction, framing their development within the broader thesis of how single-cell foundation models are reshaping biological research.
The Architecture of Single-Cell Foundation Models
Core Concepts and Model Components
Single-cell foundation models are built on the premise that biological principles can be learned from large-scale data in a self-supervised manner, analogous to how large language models learn from vast text corpora. In this paradigm, individual cells are treated as "sentences," and genes or genomic features along with their expression values are treated as "words" or tokens [74]. The core components of a typical scFM include:
- Tokenization: The process of converting raw gene expression data into discrete units for model input. Common strategies include rank-based discretization (ordering genes by expression level), bin-based discretization (grouping expression values), and value projection (continuous embeddings) [62] [74].
- Model Architecture: Most scFMs utilize transformer architectures, which employ attention mechanisms to learn relationships between genes. Some models use encoder-based architectures (e.g., BERT-like), while others use decoder-based architectures (e.g., GPT-like), with each having distinct strengths for classification versus generation tasks [74].
- Pretraining: Models are trained on massive, diverse collections of single-cell data using self-supervised objectives, most commonly masked gene modeling (MGM), where the model learns to predict randomly masked genes based on the context of other genes in the cell [74] [51].
Model Architectures and Their Biological Interpretations
The following diagram illustrates the conceptual workflow of how single-cell data is processed by foundation models, from tokenization to the generation of latent representations for downstream tasks.
Performance Across Core Analytical Tasks
Cell Type Annotation
Cell type annotation is a fundamental step in scRNA-seq analysis where cells are labeled based on their transcriptomic profiles. Traditional methods rely on manual expert knowledge or reference datasets, creating a significant bottleneck in large-scale studies.
Experimental Protocols and Methodologies
The standard protocol for benchmarking scFMs in cell type annotation involves multiple stages. First, datasets with high-quality manual annotations are selected, such as the Tabula Sapiens atlas. Data preprocessing includes normalization, log-transformation, highly variable gene selection, dimensionality reduction, and clustering using algorithms like Leiden. Differentially expressed genes are computed for each cluster, and these gene lists serve as input to foundation models for annotation [75]. Performance evaluation employs multiple metrics: direct string comparison with manual labels, Cohen's kappa (κ) for agreement, and LLM-derived ratings where models assess label matching quality (perfect, partial, or not-matching) [75]. Novel ontology-informed metrics such as scGraph-OntoRWR measure the consistency of cell type relationships captured by scFMs with prior biological knowledge, while the Lowest Common Ancestor Distance (LCAD) metric assesses the ontological proximity between misclassified cell types to gauge error severity [51].
Performance Comparison and Key Insights
Table 1: Performance of Cell Annotation Methods
| Model/Method | Annotation Accuracy (%) | Agreement with Manual Annotation (κ) | Key Strengths | Limitations |
|---|---|---|---|---|
| Claude 3.5 Sonnet | >80-90 (major types) | High | Highest agreement with manual annotation [75] | Commercial API dependency |
| scBERT | Variable across cell types | Moderate | Large-scale pretrained for cell annotation [74] | May underperform on rare cell types |
| scGPT | Variable across cell types | Moderate | Multi-omics capability [76] [74] | Computational intensity |
| AnnDictionary | Tissue-dependent | Moderate | Unified framework, multiple LLM backends [75] | Requires cluster preprocessing |
| Traditional ML | Dataset-specific | Variable | Efficient with limited resources [76] | Limited biological interpretability |
Benchmarking studies reveal that LLMs like Claude 3.5 Sonnet achieve high accuracy (>80-90%) for major cell types but performance varies significantly across models and cell types [75]. A critical finding is that no single scFM consistently outperforms all others across every task and dataset, emphasizing that model selection must be tailored to specific use cases [76] [51]. The emerging approach of using LLMs as automated annotators through tools like AnnDictionary demonstrates particular promise, achieving high agreement with manual annotations while offering scalability to atlas-sized data [75].
Batch Integration
Batch integration aligns cells across different experiments to remove technical variations while preserving biological signals. This is crucial for combining datasets from different laboratories, protocols, or platforms.
Experimental Protocols and Methodologies
Batch integration benchmarks typically involve datasets with known batch effects where the true biological variation is established. The standard workflow begins with preprocessing (normalization, highly variable gene selection) followed by application of integration methods. Performance evaluation uses two categories of metrics: (1) Batch mixing metrics such as iLISI (integration Local Inverse Simpson's Index), BatchKL (Kullback-Leibler divergence), and ASWbatch (batch silhouette width) assess how well batches are mixed; (2) Biological preservation metrics including ARI (Adjusted Rand Index), NMI (Normalized Mutual Information), and ASWcelltype evaluate how well biological cell types remain distinct after integration [77]. More advanced methods like CellANOVA utilize a "pool-of-controls" design concept to separate unwanted variation from biological variation of interest, allowing recovery of subtle biological signals erased during aggressive integration [78].
Performance Comparison and Key Insights
Table 2: Performance of Batch Integration Methods
| Model/Method | Batch Mixing (iLISI) | Biological Preservation (ASW_celltype) | Computational Efficiency | Key Innovations |
|---|---|---|---|---|
| scDML | High | High | Low memory usage | Deep metric learning, preserves rare cells [77] |
| CellANOVA | Moderate | Very High | Scalable | Recovers biological signals post-integration [78] |
| GeneMamba | High | High | Very High (linear complexity) | State-space model, efficient architecture [62] |
| scGPT | Moderate | Moderate | Moderate | Foundation model approach [76] |
| Harmony | High | Moderate | High | Rapid integration, recommended first attempt [77] |
| scVI | Moderate | Moderate | Low | Probabilistic modeling, denoising [77] |
A key insight from benchmarking is the trade-off between batch mixing and biological signal preservation. Some methods over-correct, removing biologically meaningful variation along with technical artifacts [78]. Methods like scDML excel at preserving rare cell types that might be lost by other approaches, while CellANOVA specifically addresses the recovery of biological signals erased during integration [78] [77]. Emerging architectures like GeneMamba demonstrate that state-space models can achieve competitive performance with significantly improved computational efficiency (linear vs. quadratic complexity) compared to transformer-based approaches [62].
Clinical Prediction
Clinical prediction involves using single-cell data to forecast disease outcomes, drug responses, or therapeutic efficacy, bridging the gap between basic research and clinical applications.
Experimental Protocols and Methodologies
The evaluation of scFMs for clinical prediction involves distinct experimental designs depending on the clinical context. For drug sensitivity prediction, models are typically trained on single-cell data from cell lines or patient-derived samples treated with various compounds, then tested on held-out datasets to predict response metrics [76] [51]. For cancer cell identification, models are benchmarked on their ability to distinguish malignant from non-malignant cells across multiple cancer types, with performance measured via AUC-ROC, precision-recall, and related classification metrics [76] [51]. For biomarker discovery, methods are evaluated by their capacity to identify genes or gene signatures that predict clinical outcomes, with validation against established clinical biomarkers or orthogonal assays [79] [80].
Performance Comparison and Key Insights
Table 3: Performance of Clinical Prediction Methods
| Model/Method | Drug Sensitivity Prediction | Cancer Cell Identification | Biomarker Discovery | Clinical Applicability |
|---|---|---|---|---|
| scFoundation | High across 4 drugs [76] | Variable across 7 cancer types [76] | Moderate | High potential |
| Geneformer | Moderate | High in specific cancers [51] | High | Demonstrated in cardiomyopathy [51] |
| scGPT | Moderate | Moderate | Moderate | Multi-omics advantage |
| Traditional ML | Dataset-specific | Dataset-specific | Limited | Well-established but limited scope |
Benchmarking across seven cancer types and four drugs reveals that scFMs show promise in clinical applications but with significant variability across cancer types and compounds [76]. A notable finding is that cell-type specific expression in disease-relevant tissues is a robust predictor of a drug target's progression from Phase I to Phase II clinical trials, highlighting the clinical relevance of single-cell resolution [79]. However, in some scenarios, simpler machine learning models can outperform foundation models, particularly when training data is limited or computational resources are constrained [76] [51].
Table 4: Essential Computational Tools for Single-Cell Foundation Model Research
| Tool/Resource | Function | Key Features | Access |
|---|---|---|---|
| AnnDictionary | LLM-based cell type annotation | Multi-LLM backend, parallel processing, minimal code configuration [75] | https://github.com/ggit12/anndictionary/ |
| scDML | Batch integration | Deep metric learning, rare cell type preservation [77] | https://github.com/eleozzr/scDML |
| CellANOVA | Signal recovery post-integration | Pool-of-controls design, recovers biological variation [78] | Statistical model/R package |
| GeneMamba | Efficient foundation modeling | State-space model, linear complexity, scalable to 50M+ cells [62] | Available on arXiv |
| Scanpy | Single-cell analysis ecosystem | Standard preprocessing, integration, and visualization [77] | Python package |
| CZ CELLxGENE | Data repository | Curated single-cell datasets for pretraining [74] | https://cellxgene.cziscience.com/ |
Integrated Workflow for Single-Cell Analysis
The following diagram illustrates how the various tools and methods can be combined into a comprehensive workflow for single-cell analysis, from raw data processing to biological insights.
Single-cell foundation models represent a paradigm shift in how we analyze and interpret single-cell transcriptomic data. Rather than treating each analysis as a discrete problem, scFMs leverage patterns learned from millions of cells to provide robust, context-aware solutions across diverse applications. The benchmarking evidence clearly indicates that while these models show remarkable versatility, their performance is highly task-dependent. For cell type annotation, LLM-based approaches like AnnDictionary with Claude 3.5 Sonnet backend demonstrate exceptional accuracy. For batch integration, methods like scDML and CellANOVA excel at preserving biological signals while removing technical artifacts. For clinical prediction, foundation models show promise but face greater variability across diseases and compounds.
The broader thesis emerging from this research is that single-cell foundation models work by learning fundamental biological principles encoded in transcriptomic patterns across diverse cell types, states, and conditions. This learned representation captures intrinsic properties of cellular identity and function that transfer effectively to downstream tasks. However, model selection must be guided by specific analytical needs, dataset characteristics, and computational constraints. As these models continue to evolve, they hold tremendous potential to accelerate drug discovery, enhance our understanding of disease mechanisms, and ultimately bridge the gap between single-cell genomics and clinical applications.
The advent of single-cell foundation models (scFMs) represents a paradigm shift in computational biology, bringing artificial intelligence directly into cell biology [1]. These large-scale deep learning models, pretrained on vast single-cell datasets, promise to revolutionize how we interpret cellular heterogeneity and complex regulatory networks. However, their powerful capabilities introduce a critical challenge for researchers: the need to balance technical metrics—quantitative measures of model performance and data structure preservation—with biological relevance—the actual biological insights and mechanisms these models can uncover [1] [3]. This balancing act is not merely academic; it determines whether scFMs will transition from computational marvels to indispensable tools in biomedical research and therapeutic development.
This challenge emerges from the fundamental nature of single-cell data itself. Single-cell RNA sequencing (scRNA-seq) data exhibit characteristics of high sparsity, high dimensionality, and low signal-to-noise ratio [3]. When applying dimensionality reduction techniques or foundation models to such data, technical artifacts can easily be mistaken for biological signals, or conversely, subtle but biologically important patterns can be obscured by technical variation [81]. For researchers and drug development professionals, this balance carries high stakes: misinterpretations can lead to erroneous biological conclusions or missed therapeutic opportunities.
The Evaluation Framework: Technical and Biological Dimensions
Technical Metrics: Quantifying Data Structure Preservation
Technical metrics primarily focus on how well computational methods preserve the inherent structure of high-dimensional single-cell data during transformation to lower-dimensional embeddings. These metrics provide essential quantitative standards for evaluating dimensionality reduction techniques and foundation model outputs.
Table 1: Core Technical Metrics for Evaluating Single-Cell Data Transformations
| Metric Category | Specific Metric | Interpretation | Calculation Basis |
|---|---|---|---|
| Global Structure | Earth-Mover's Distance (EMD) | Quantifies structural alteration of cell distance distribution | Energy cost of shifting native distribution to latent distribution [81] |
| Global Structure | Distance Correlation | Measures preservation of unique cell-cell distances | Pearson correlation of native vs. latent space distances [81] |
| Local Structure | K-Nearest Neighbor (KNN) Preservation | Percentage of local neighborhoods conserved | Binary matrix comparison of KNN graphs [81] |
| Batch Effects | Batch Integration Scores | Separation of batches vs. biological groups | Multiple metrics evaluating batch mixing and biological conservation [82] |
The foundation of these technical assessments begins with understanding cell-cell distances in native high-dimensional space. In scRNA-seq, counts of unique molecular identifiers (UMIs) for each gene constitute the features, with every observation representing a single cell, forming an m × n matrix (observations × features) [81]. Global data structure is constructed by calculating an m × m matrix containing pairwise distances between all observations, from which a probability density distribution can be derived [81]. Local "neighborhoods" are defined via K nearest-neighbor (KNN) graphs, represented as binary m × m matrices that define the K cells with shortest distances to each cell [81]. These native space relationships serve as the benchmark against which transformed data are compared.
Different technical metrics reveal different aspects of preservation performance. For instance, uniform manifold approximation and projection (UMAP) tends to compress small, local distances more significantly than t-distributed stochastic neighbor embedding (t-SNE), while both methods maintain relative global structure as indicated by favorable correlation of large distances [81]. This compression characteristic in UMAP embeddings, while producing visually condensed clusters that may be easier to interpret, results in greater information loss reflected in less favorable preservation metrics [81]. Understanding these method-specific characteristics is essential for proper interpretation of results.
Biologically Grounded Evaluation: Beyond Technical Metrics
While technical metrics provide essential quantitative benchmarks, they fall short in capturing the biological plausibility and relevance of scFM outputs. biologically grounded evaluation requires different approaches that connect computational outputs to established biological knowledge.
Table 2: Biologically Informed Evaluation Metrics for scFMs
| Evaluation Approach | Specific Metric | Biological Basis | Application Context |
|---|---|---|---|
| Cell Ontology-Informed | scGraph-OntoRWR | Consistency of cell type relationships with prior knowledge | Measures if model captures known biological relationships between cell types [3] |
| Cell Ontology-Informed | Lowest Common Ancestor Distance (LCAD) | Ontological proximity between misclassified types | Assesses severity of annotation errors in biological context [3] |
| Gene Functional | GO Term Prediction | Association of gene embeddings with biological processes | Tests if functionally related genes cluster in embedding space [3] |
| Tissue Specificity | Tissue Specificity Prediction | Linkage of gene embeddings to tissue context | Evaluates biological contextualization of gene representations [3] |
The scGraph-OntoRWR metric represents a particularly innovative approach to biological evaluation. Rather than simply measuring clustering accuracy or separation, it evaluates whether the relational structure between cell types captured by scFMs aligns with established biological knowledge encoded in cell ontologies [3]. Similarly, the LCAD metric recognizes that not all cell type misclassifications are equally serious—confusing closely related cell types is less concerning than confusing biologically distant types, and this metric quantifies this biological nuance [3].
These biologically informed metrics address a critical gap in traditional evaluation frameworks. A model might achieve excellent technical metrics for batch integration or clustering while still producing biologically misleading representations. For instance, a method might over-integrate batches, removing not just technical artifacts but genuine biological variation, such as subtle but meaningful differences between patient subgroups or disease states [82]. Only evaluation approaches that incorporate biological ground truth can detect such failures.
Figure 1: Dual-Path Evaluation Framework for Single-Cell Foundation Models. This workflow illustrates the parallel assessment of technical and biological dimensions required for comprehensive model interpretation.
Integrated Workflow for Balanced Evaluation
Experimental Protocol for Comprehensive scFM Assessment
Implementing a balanced evaluation requires a structured experimental approach that systematically addresses both technical and biological dimensions. The following protocol outlines key steps for comprehensive scFM assessment:
Data Preparation and Quality Control Begin with raw count matrices from single-cell technologies (e.g., droplet-based scRNA-seq). Conduct rigorous quality control to filter low-quality cells by setting thresholds for detected genes, count depth, and mitochondrial count fraction [82]. Address ambient RNA contamination using specialized tools such as SoupX or CellBender, and detect doublets with scDblFinder, which has demonstrated superior performance in benchmarks [82]. Normalize data using appropriate methods—Scran normalization for batch correction tasks or analytical Pearson residuals for biologically variable gene selection [82].
Technical Benchmarking Apply scFMs to obtain low-dimensional embeddings. Calculate global preservation metrics (EMD, distance correlation) comparing native high-dimensional space to scFM-derived latent space [81]. Evaluate local structure preservation through KNN graph conservation percentages. Assess batch integration using specialized metrics that separately quantify batch mixing and biological conservation, potentially employing the scIB package which implements comprehensive benchmarking metrics [82].
Biological Validation Extract gene and cell embeddings from scFMs. For gene-level validation, evaluate whether embeddings capture functional relationships by testing their performance in predicting Gene Ontology terms and tissue specificity [3]. For cell-level validation, apply ontology-informed metrics including scGraph-OntoRWR to measure consistency with known cell type relationships and LCAD to assess biological plausibility of misclassifications [3]. Incorporate challenging biological scenarios such as novel cell types, cross-tissue homogeneity, and intra-tumor heterogeneity to stress-test model performance.
Iterative Refinement and Interpretation Analyze discrepancies between technical and biological metrics. Poor technical metrics with strong biological insights may indicate issues with metric selection or calculation, while strong technical metrics with poor biological relevance may signal overfitting or loss of biologically meaningful variation. Use the roughness index (ROGI) as a proxy to evaluate the suitability of different models for specific datasets and tasks [3].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Computational Tools for scFM Evaluation
| Tool Category | Specific Tools | Primary Function | Application Context |
|---|---|---|---|
| Foundation Models | scGPT, Geneformer, scBERT | Large-scale pretraining on single-cell data | Base models for transfer learning and embedding extraction [1] [3] |
| Integration Methods | Harmony, scANVI, scVI | Batch correction and data integration | Removing technical variation while preserving biological signals [82] |
| Evaluation Frameworks | scIB, scGraph-OntoRWR | Comprehensive metric calculation | Quantifying technical and biological performance [3] [82] |
| Visualization Platforms | CZ CELLxGENE, UCSC Cell Browser | Data exploration and result interpretation | Interactive visualization of single-cell data and annotations [1] |
Discussion and Future Directions
Practical Implications for Research and Drug Development
The balanced evaluation framework presented here has significant implications for both basic research and translational applications. For researchers constructing cell atlases, over-reliance on technical metrics alone might produce beautifully integrated maps that nevertheless obscure biologically important rare cell populations or subtle transitional states. Conversely, dismissing technical metrics in favor of purely biological assessment risks being misled by technical artifacts that resemble biological patterns.
In drug development contexts, where scFMs are increasingly applied to tasks like cancer cell identification and drug sensitivity prediction [3], the stakes for balanced evaluation are particularly high. A model with excellent technical metrics for batch integration might inadvertently remove patient-specific variation that is crucial for predicting differential treatment response. Similarly, in tumor microenvironment studies, the ability to distinguish biologically distinct but transcriptionally similar cell states could have significant therapeutic implications, making biologically informed evaluation essential.
Emerging Challenges and Opportunities
Despite recent progress, several challenges remain in balancing technical and biological evaluation of scFMs. First, the field still lacks consensus on optimal tokenization strategies for representing single-cell data in foundation models [1] [22]. Genes lack natural sequential ordering unlike words in language, forcing researchers to adopt various strategies such as ranking by expression levels or binning by expression values [1]. How these different tokenization approaches affect the biological plausibility of model outputs remains incompletely understood.
Second, current benchmarking reveals that no single scFM consistently outperforms others across all tasks [3]. This underscores the importance of task-specific model selection rather than seeking a universal best model. Factors such as dataset size, task complexity, need for biological interpretability, and computational resources should guide model selection [3].
Finally, as scFMs increasingly incorporate multiple modalities—including chromatin accessibility, spatial information, and protein expression [1] [82]—evaluation frameworks must evolve to assess multimodal integration. This will require novel metrics that can evaluate not just how well each modality is represented, but how effectively models capture biologically meaningful interactions between modalities.
Balancing technical metrics with biological relevance is not merely an academic exercise but a practical necessity for realizing the potential of single-cell foundation models. Technical metrics provide essential quantitative rigor, ensuring that data transformations faithfully preserve underlying structures, while biological evaluation grounds computational outputs in physiological reality. The most insightful applications of scFMs will emerge from frameworks that honor both dimensions, using technical metrics as necessary checkpoints rather than final endpoints, and biological relevance as the ultimate criterion for success. As these models continue to evolve, maintaining this dual focus will be crucial for translating computational advances into genuine biological insights and therapeutic breakthroughs.
Conclusion
Single-cell foundation models represent a powerful paradigm shift in computational biology, offering unprecedented scale and versatility for analyzing cellular systems. While they show remarkable potential for tasks ranging from cell type annotation to drug sensitivity prediction, current implementations face significant challenges in zero-shot performance and don't consistently outperform simpler traditional methods. The field is rapidly evolving, with scaling experiments showing that performance improves predictably with both data volume and parameter count. Future success will depend on developing more biologically intuitive model architectures, creating standardized evaluation frameworks, and building user-friendly interfaces that make these powerful tools accessible to the broader research community. As models continue to scale and incorporate multimodal data, scFMs are poised to become indispensable tools for unlocking deeper insights into cellular function, disease mechanisms, and personalized therapeutic development.
References
- 1. Single-cell foundation models: bringing artificial ... [https://www.nature.com/articles/s12276-025-01547-5]
- 2. Single-cell foundation models: The next big thing? [https://frontlinegenomics.com/single-cell-foundational-models-the-next-big-thing/]
- 3. Biology-driven insights into the power of single-cell foundation ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03781-6]
- 4. General-purpose pre-trained large cellular models for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11565237/]
- 5. Foundation models in molecular biology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11252241/]
- 6. Foundation Model: A New Era for Plant Single-cell Genomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC12380448/]
- 7. [2210.12385] Deep Learning in Single-Cell Analysis [https://arxiv.org/abs/2210.12385]
- 8. New Foundation Model Reveals How Cells Are Organized ... [https://www.helmholtz-munich.de/en/newsroom/news-all/artikel/new-foundation-model-reveals-how-cells-are-organized-in-tissues]
- 9. Transformative advances in single-cell omics - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12560279/]
- 10. Nicheformer: a foundation model for single-cell and spatial ... [https://www.nature.com/articles/s41592-025-02814-z]
- 11. scViewer: An Interactive Single-Cell Gene Expression ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10253102/]
- 12. Teddy: A family of foundation models for understanding ... [https://arxiv.org/html/2503.03485v1]
- 13. CZ CELLxGENE Discover - Cellular Visualization Tool [https://cellxgene.cziscience.com/]
- 14. Public Single-Cell RNA-Seq Databases: A Comprehensive ... [https://www.nygen.io/resources/blog/public-single-cell-rna-seq-databases]
- 15. Census Models [https://cellxgene.cziscience.com/census-models]
- 16. The future of rapid and automated single-cell data analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11184658/]
- 17. Multimodal learning enables chat-based exploration of ... [https://www.nature.com/articles/s41587-025-02857-9]
- 18. Benchmarking deep learning methods for ... - Genome Biology [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03869-z]
- 19. Single-cell analyses reveal increased gene expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081229/]
- 20. Interpretable Multi-task Analysis of Single-Cell RNA-seq ... [https://pubmed.ncbi.nlm.nih.gov/41023370/]
- 21. Benchmarking integration of single-cell differential ... [https://www.nature.com/articles/s41467-023-37126-3]
- 22. A Survey on Foundation Language Models for Single-cell ... [https://aclanthology.org/2025.acl-long.26/]
- 23. Article BioLLM: A standardized framework for integrating ... [https://www.sciencedirect.com/science/article/pii/S2666389925001746]
- 24. GeneRAIN: multifaceted representation of genes via deep ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03749-6]
- 25. mRNABERT: advancing mRNA sequence design with a ... [https://www.nature.com/articles/s41467-025-65340-8]
- 26. Effect of tokenization on transformers for biological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11055402/]
- 27. Tokenization and deep learning architectures in genomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC12356405/]
- 28. What is Positional Encoding? | IBM [https://www.ibm.com/think/topics/positional-encoding]
- 29. A Gentle Introduction to Positional Encoding in Transformer ... [https://www.machinelearningmastery.com/a-gentle-introduction-to-positional-encoding-in-transformer-models-part-1/]
- 30. Positional Encoding Explained: A Deep Dive into ... [https://medium.com/thedeephub/positional-encoding-explained-a-deep-dive-into-transformer-pe-65cfe8cfe10b]
- 31. Positional Encoding in Transformers [https://www.geeksforgeeks.org/nlp/positional-encoding-in-transformers/]
- 32. GenAI Concept — Positional Encoding [https://medium.com/@shuv.sdr/genai-concept-positional-encoding-23617018581e]
- 33. A Survey from the Perspective of Positional Encoding [https://arxiv.org/html/2312.17044v4]
- 34. CellFM: a large-scale foundation model pre-trained on transcriptomics of 100 million human cells | Nature Communications [https://www.nature.com/articles/s41467-025-59926-5]
- 35. Positional encoding-guided transformer-based multiple ... [https://www.sciencedirect.com/science/article/abs/pii/S016926072400484X]
- 36. Delineating the effective use of self-supervised learning in ... [https://www.nature.com/articles/s42256-024-00934-3]
- 37. Single-cell foundation models: bringing artificial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12586647/]
- 38. CellFM: a large-scale foundation model pre-trained on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12092794/]
- 39. scMapNet: Marker-based cell type annotation of scRNA- ... [https://www.sciencedirect.com/science/article/pii/S2090123225008501]
- 40. Robust self-supervised machine learning for single cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12157554/]
- 41. guidelines for annotating single-cell transcriptomic maps ... [https://pubmed.ncbi.nlm.nih.gov/34031612/]
- 42. Using LLMs and Explainable ML to Analyze Biomarkers at ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10605495/]
- 43. A drug response prediction method for single-cell tumors ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2025.1631898/full]
- 44. Deep learning-driven drug response prediction and ... [https://www.nature.com/articles/s41598-025-91571-2]
- 45. [2503.03485] TEDDY : A Family Of Foundation For... Models [https://arxiv.org/abs/2503.03485]
- 46. scPlantLLM: A Foundation Model for Exploring Single-cell ... [https://pubmed.ncbi.nlm.nih.gov/40094454/]
- 47. Merck/ TEDDY · Hugging Face [https://huggingface.co/Merck/TEDDY]
- 48. Solution Building: Merck Avoids One-Size-Fits-All Approach to AI and ML [https://www.genengnews.com/topics/artificial-intelligence/solution-building-merck-avoids-one-size-fits-all-approach-to-ai-and-ml/]
- 49. Why simple models outperformed deep learning in gene ... [https://www.linkedin.com/posts/vsavova_deep-learning-based-gene-perturbation-effect-activity-7360314175551639552-mjBK]
- 50. scDrugMap: Benchmarking Large Foundation Models for ... [https://arxiv.org/abs/2505.05612]
- 51. Biology-driven insights into the power of single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492631/]
- 52. Benchmarking deep learning methods for biologically ... [https://pubmed.ncbi.nlm.nih.gov/41267095/]
- 53. 江瑞教授团队提出国际上首个单细胞表观基因组基础大模型 ... [https://www.au.tsinghua.edu.cn/info/1133/4246.htm]
- 54. Transformative advances in single-cell omics: a ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-07091-0]
- 55. Zero-shot evaluation reveals limitations of single-cell ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03574-x]
- 56. Foundation models in drug discovery: phenomenal growth ... [https://pubmed.ncbi.nlm.nih.gov/41167387/]
- 57. Zero-shot evaluation reveals limitations of foundation ... [https://www.microsoft.com/en-us/research/articles/zero-shot-evaluation-reveals-limitations-of-foundation-models-in-single-cell-biology/]
- 58. Zero-Shot Learning: A Practical Guide [https://www.lightly.ai/blog/zero-shot-learning]
- 59. : A standardized BioLLM for integrating and benchmarking... framework [https://pubmed.ncbi.nlm.nih.gov/40843339/]
- 60. BioLLM: A standardized framework for integrating and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12365531/]
- 61. GitHub - BGIResearch/ BioLLM [https://github.com/BGIResearch/BioLLM]
- 62. Efficient and Effective Foundation Model on Single Cell Data [https://arxiv.org/html/2504.16956v1]
- 63. scKGBERT: a knowledge-enhanced foundation model for ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03862-6]
- 64. From Data to Drugs: The Role of Artificial Intelligence in Drug ... [https://wyss.harvard.edu/news/from-data-to-drugs-the-role-of-artificial-intelligence-in-drug-discovery/]
- 65. The future of biological foundation models and value ... [https://www.scalevp.com/insights/the-future-of-biological-foundation-models-and-value-creation-in-ai-driven-drug-discovery/]
- 66. Benchmarking foundation cell models for post-perturbation ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11600-2]
- 67. Closing the loop: Teaching single-cell foundation models to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12265564/]
- 68. Transcoder-based Circuit Analysis for Interpretable Single- ... [https://arxiv.org/html/2509.14723v1]
- 69. Parameter-Efficient Fine-Tuning Enhances Adaptation of Single Cell... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10862733/]
- 70. Deep-learning-based gene perturbation effect prediction ... [https://www.nature.com/articles/s41592-025-02772-6]
- 71. GitHub - HelloWorldLTY/scEval: Codes for paper: Evaluating the... [https://github.com/HelloWorldLTY/scEval]
- 72. The Invisible Framework Organizing Life: Biological Ontologies [https://www.biochemicalsci.com/posts/the-invisible-framework-organizing-life-biological-ontologies]
- 73. PAST: A multimodal single-cell foundation model for ... [https://arxiv.org/abs/2507.06418]
- 74. - Single : bringing artificial intelligence into cell ... foundation models cell [https://www.nature.com/articles/s12276-025-01547-5?error=cookies_not_supported&code=a5069175-7780-4c9c-9dd1-1958ed8f1041]
- 75. Benchmarking cell type and gene set annotation by large ... [https://www.nature.com/articles/s41467-025-64511-x]
- 76. Biology-driven insights into the power of single - cell foundation models [https://pubmed.ncbi.nlm.nih.gov/41044630/]
- 77. Batch alignment of single-cell transcriptomics data using ... [https://www.nature.com/articles/s41467-023-36635-5]
- 78. Signal recovery in single cell batch integration - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC10197537/]
- 79. Single Cell Sequencing and The Future of Drug Discovery [https://www.parsebiosciences.com/blog/single-cell-sequencing-and-the-future-of-drug-discovery/]
- 80. Single-cell technology for drug discovery and development [https://www.frontiersin.org/journals/drug-discovery/articles/10.3389/fddsv.2024.1459962/full]
- 81. A Quantitative Framework for Evaluating Single-Cell Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7305633/]
- 82. Best practices for single-cell analysis across modalities [https://pmc.ncbi.nlm.nih.gov/articles/PMC10066026/]